
长双歧杆菌A17 胞外多糖合成基因簇的分析
2023-11-09 12:28:14张文丽呼鑫荣旭日花
食品与生物技术学报 2023年10期
张文丽, 王 英, 石 莹, 呼鑫荣, 旭日花
(内蒙古大学生命科学学院, 内蒙古呼和浩特 010021)
双歧杆菌(Bifidobacterium)是一种高G+C 含量的厌氧革兰氏阳性杆状菌,属放线菌门[1],1899 年由Tissier 首次从婴儿粪便中分离得到[2]。作为人出生后肠道内最早的细菌定植者之一[3],其数量和组成随时间和年龄而变化[4-5]。 根据不同年龄段人肠道内存在的菌种不同可将双歧杆菌分为: 婴儿双歧杆菌(Bifidobacterium infantis)、 青春双歧杆菌(B.adolescentis)、长双歧杆菌(B. longum)等[6]。双歧杆菌作为一种益生菌,被认为主要通过提供营养和非营养的化合物来促进宿主新陈代谢进而维护宿主的健康[7]。 同时双歧杆菌的其他积极作用表现在缓解肠道疾病方面,例如肠易激综合征(IBS)和炎症性肠病(IBD)等[8-9]。 双歧杆菌EPS 是双歧杆菌在生长代谢过程中分泌的一类长链聚合物,根据其相对于菌体的位置分为荚膜多糖和黏液多糖。 因EPS 具有抑菌、抗氧化、抗肿瘤、免疫调节等多种生物活性,因此,能够产生EPS 的双歧杆菌在相关益生菌研究中具有重要的应用价值和广阔的应用前景。
胞外多糖(exopolysaccharides,EPS)是一种存在于大多数细菌中的碳水化合聚合物,它们可以松散附着在细菌细胞表面或释放到周围的细胞环境,既作为细菌保护性表层,也与周围环境发生相互作用[10]。多年来,对细菌胞外多糖研究的热度一直有增无减,不仅仅因为它们对生物膜形成的贡献,更重要的是因为EPS 是几种传染病的潜在的毒力因子[11]。 近几年由于发现EPS 能够调节与周围环境的交流以及它们对宿主健康维护的贡献及其在食品、化妆品和医药方面的潜在应用使得对EPS 的研究受到更大的关注[11]。
EPS 合成分为糖核苷酸合成和eps 基因簇合成2 个阶段。糖类经转运系统由细胞外转运到细胞内,再经一系列酶促反应生成糖核苷酸。 糖核苷酸作为合成EPS 的前体物质, 在pgmA、galE、galU等管家基因和eps基因簇的调控下完成EPS 的合成、聚合以及向细胞外输出等[12]。 在乳酸菌菌株中指导EPS合成的基因首次在Streptococcus thermophilusSfi6被报道,该簇长为14.5 kb,由13 个基因组成[13]。 据报道, 乳酸菌EPS 生物合成过程中各前体物质和酶,均已被证明能够影响EPS 的产量[14]。 其中Lactococcus lactisstrain NIZO B40 菌株含有一个42.2 kb EPS 质粒,存在一个12 kbeps操纵子。将整个NIZO B40eps基因簇克隆到高拷贝数载体pIL253 中, 导致EPS 生产水平几乎增加了4 倍[15]。然而,eps基因簇并不是影响EPS 生物合成的唯一决定性因素。 Welman 等人研究表明6-磷酸葡萄糖似乎是连接EPS 合成代谢途径和糖酵解分解代谢途径的中间关键体[16]。 这些结果表明EPS 生物合成机制在不同的菌株中是不同的、复杂的。
目前,相较于乳酸菌的其他种属,双歧杆菌EPS合成相关基因及其生物工程改造方面的研究报道较少。 因此,作者阐明EPS 前体糖核苷酸合成途径并寻找与EPS 合成相关的基因簇, 在此基础上将EPS 合成相关的基因进行异源表达, 为进一步探索EPS 合成途径提供理论基础。
1 材料与方法
1.1 材料与试剂
长双歧杆菌A17:分离自健康婴儿粪便,保存于内蒙古大学生命科学学院;pET-28a 质粒:北纳生物颂司产品; 大肠杆菌感受态细胞DH5α:Trans Gen Biotech 产品;MRS 培养基: 北京陆桥公司产品;溶菌酶:天根生化公司产品;细菌基因组DNA 提取试剂盒: 天根生化公司产品;Quick DNA Purification Kit DNA 产物纯化试剂盒、Plasmid Mini Kit 质粒小提试剂盒、Gel Extraction Kit 胶回收试剂盒:CWBIO公司产品;T4 DNA Ligase:TaKaRa 公司产品。
1.2 仪器
超净工作台:南京MDS 公司产品;LRH-250 型生化培养箱:上海一恒科学仪器公司产品;YU-1810型紫外可见分光光度计:北京普析通用仪器公司产品; 琼脂糖凝胶电泳仪:BIO-RAD 公司产品;PCR扩增仪:德国Jena 分析仪器公司产品;核酸浓度检测仪:Themro 公司产品。
1.3 研究方法
1.3.1 生物信息学分析使用基于IlluminaMiSeq测序平台的二代测序技术和基于PacBio 测序平台的三代测序技术对文库进行测序后, 采用NCBI 公认的原核生物编码基因预测软件GeneMarkS 对所有基因进行预测。 最后使用eggNOG-mapper、KAAS自动化注释系统、InterPro、BLASTP 等软件进行生物信息学分析。
1.3.2 重组表达载体的构建
1) PCR 引物设计 根据NCBI 数据库中长双歧杆菌pgm基因完整序列,设计引物并交由华大科技公司合成(见表1)。 PCR(25 μL 体系)反应条件:95 ℃预变性3 min、95 ℃变性30 s、57 ℃退火30 s、72 ℃延伸2 min,共35 个循环,72 ℃终延伸6 min。PCR 扩增后电泳检测,并进行胶回收、纯化。

表1 PCR 扩增所用引物及其序列Table 1 Primers and their sequences used for PCR amplification
2) pET28a-pgm重组表达载体的构建 用质粒提取试剂盒从大肠杆菌中提取pET-28a 质粒,提取的质粒和克隆后测序正确的pgm基因分别用BamHⅠ和Hind III 酶双酶切, 酶切后的混合物用Quick DNA Purification Kit DNA 产物纯化试剂盒纯化,16℃连接14 h。 连接体系(10 μL ):1 μL 10×T4 连接缓冲液,1 μL T4 DNA 连接酶,1 μL 双酶切后的pET28a 载体,3 μL 双酶切后的pgm基因,4 μL ddH2O。
3) 重组质粒pET28a-pgm的转化与鉴定 取50 μL DH5α 与重组质粒混合,冰上放置30 min,42℃水浴热击90 s,迅速置于冰上冷却5 min,加入1 mL LB 液体培养基(不含Kan)中,37 ℃培养1 h 完成转化。 转化完成的菌液涂布于LB 固体平板上(含Kan),37 ℃培养18 h 筛选阳性重组子。 随机挑取4个阳性重组子单菌落进行菌落PCR。 将挑取的单菌落悬浮于20 μL 质量分数10%的TritonX-100 细胞裂解液中,置于沸水浴3 min,使细胞裂解,以裂解液为模板,进行PCR 并电泳检测。 挑取电泳条带正确的菌落PCR 的副板单菌落于5 mL LB 液体培养基 (Kan 50 μg/mL) 中,37 ℃摇床培养12 h。 用Plasmid Mini Kit 质粒提取试剂盒提取质粒, 送测序。 同时对重组质粒进行双酶切和电泳检测,鉴定pET28a-pgm重组表达载体是否构建成功。 重组成功的大肠杆菌在甘油管中保种,-80 ℃冰箱保存。
4) 重组质粒的诱导表达 抗性平板上挑取单菌落,加入20 mL 含卡那抗性的LB 液体培养基,过夜摇菌, 以体积分数1%的接种转接于50 mL 含卡那抗性的LB 液体培养基, 待菌液的OD 值为0.6时, 加入0.4 mmol/L 的IPTG,16 ℃诱导表达12 h,使蛋白表达量达到最大。 用5 mL NTA-0 平衡缓冲液悬浮破碎菌液;10 000g,20 min 离心; 用5 mL NTA-0 重悬沉淀; 将上清和沉淀分别进行SDSPAGE 凝胶电泳。
5) 目的蛋白的分离与纯化 将上清液进行His-tag 亲和层析(Invitrogen®),层析产物浓缩后进行凝胶电泳,检测目的重组蛋白质。 层析产物进一步用AKTA Purifier 分子筛去除其他的杂蛋白质,收集特定峰所对应的样品,SDS-PAGE 凝胶电泳检测验证是否获得了高纯度目的重组蛋白质。 以牛血清白蛋白(Albumin from bovine serum,BSA)作为标准品,用Bradford 法测定目的蛋白质质量浓度。
2 结果与分析
2.1 EPS 基因簇预测结果
以已报道的双歧杆菌EPS 基因簇为基础,利用不同双歧杆菌此类簇边界处的保守区域进行分析后,发现双歧杆菌A17 存在一个由19 个基因(chr-354 到chr-413)组成的,长度约为20 kb 的簇,命名为A17eps基因簇。 EPS 基因簇结果如图1 所示。
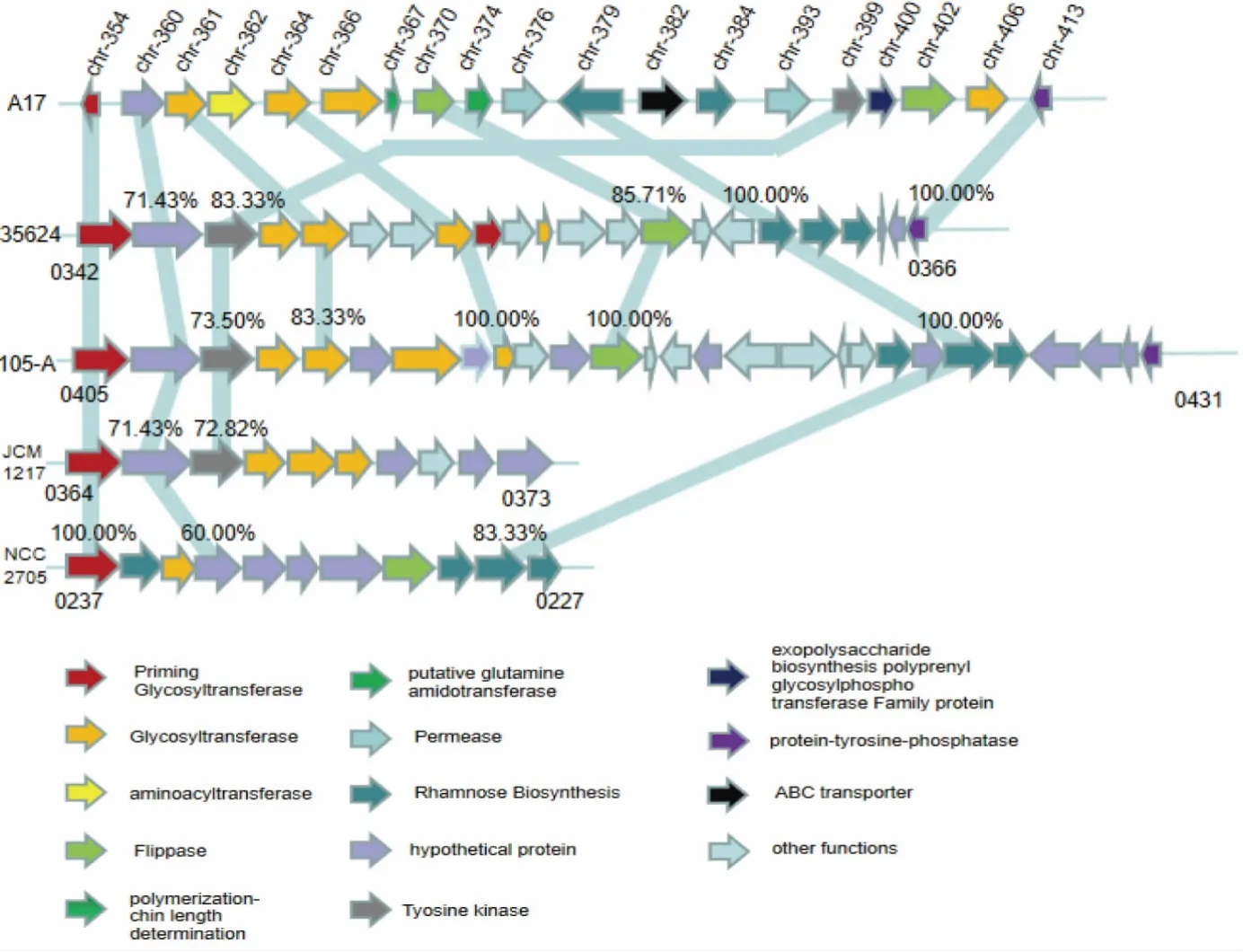
图1 A17 eps 基因簇与其他EPS 基因簇的比较分析结果Fig. 1 Comparative analysis results of A17 eps gene cluster and other EPS gene clusters
2.2 EPS 前体糖核苷酸生物合成途径分析
据图2 分析得知,乳糖可被lacZ和LCT编码的乳糖酶水解为D-半乳糖和α-D-葡萄糖,再经半乳糖激酶(galK)、葡萄糖磷酸变位酶(pgm)以及UDP-糖焦磷酸化酶 (USP) 等的作用下分别转化形成UDP-半乳糖和UDP-葡萄糖。 其中,UDP-半乳糖和UDP-葡萄糖之间可经UDP 葡萄糖-4-表异构酶(galE)相互进行转化。 经乳糖代谢产生的UDP 葡萄糖可与新的1-磷酸半乳糖反应生成1-磷酸葡萄糖,也可进入糖原合成通路参与糖原的合成。 葡萄糖经葡糖激酶(glk)转化为葡萄糖六磷酸,磷酸葡萄糖变位酶(pgm)使葡萄糖6-磷酸进一步形成葡萄糖1-磷酸, 产物再经UTP-葡萄糖-1-磷酸尿苷酰转移酶(galU) 、UDP-糖焦磷酸化酶(USP)转变最后形成UDP-葡萄糖。因此,长双歧杆菌A17 菌株利用葡萄糖, 乳糖, 通过以上方式形成2 种糖核苷酸(UDP-葡萄糖、UDP-半乳糖)。为后续合成EPS A17提供原材料,EPS A17 的结构已另文发表[20]。
2.3 pET28a-pgm 重组表达载体的构建与鉴定
pgm基因的PCR 扩增产物电泳结果如图3 所示。 菌落PCR 的扩增结果如图4 所示。
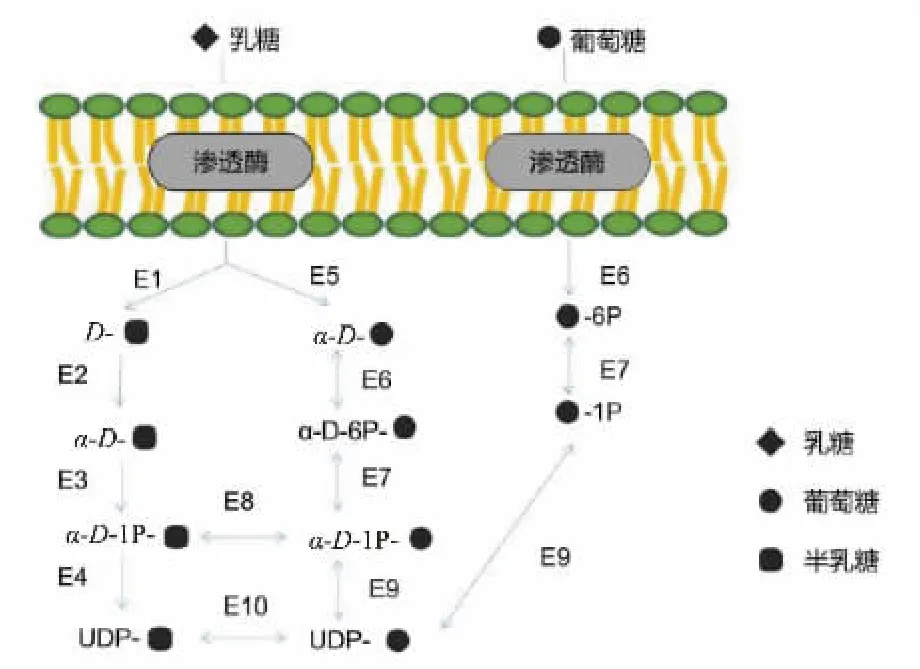
E1:β-半乳糖苷酶;E2:醛糖1-差向异构酶;E3:半乳糖激酶;E4:UDP-糖焦磷酸化酶;E5: 乳糖酶-根皮苷水解酶;E6:葡萄糖激酶;E7: 葡萄糖磷酸变位酶;E8:UDP-葡萄糖-己糖-1-磷酸尿苷酰转移酶;E9:UTP-葡萄糖-1-磷酸尿苷酰转移酶;E10:UDP-葡萄糖-4-差向异构酶。
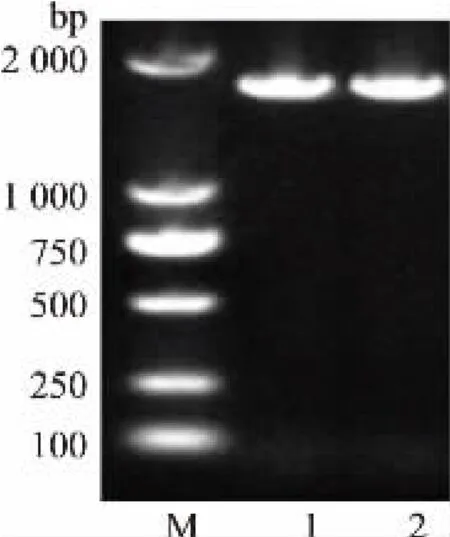
图3 pgm 基因的PCR 扩增产物电泳图Fig. 3 Electrophoresis of the PCR amplification product of gene pgm

图4 菌落PCR 的扩增电泳图Fig. 4 Electrophoresis of colony PCR amplification
电泳结果显示,基因片段大小接近1 700 bp 左右与目标片段(1 677 bp)长度吻合。 菌落PCR 结果表明该载体成功转化到大肠杆菌中。 pET28a-pgm重组表达载体构建及双酶切电泳结果如图5 所示。

图5 pET28a-pgm 重组表达载体构建及相关产物的电泳结果Fig. 5 Electrophoresis results of the pET28a-pgm recombinant expression vector constr uction and related products
在图5 中,1 号泳道为空pET-28a 质粒载体,长度为5 369 bp,但是电泳图中的克隆片段为4 000~7 000 bp, 这是由于在4 000~7 000 bp 区域中质粒超螺旋形式的比例最大, 与线状DNA 和开环DNA相比,超螺旋DNA 电泳速度最快,因此4 000~5 000 bp 出现最亮的条带。 2 号泳道为空载体双酶切产物,酶切后电泳速度要比原载体慢,故在5 000~6 000 bp 出现最亮的条带。 3、4 号泳道的扩增片段长度大约7 000 bp 左右,与pET28a-pgm重组表达载体序列长度的6 974 bp 吻合。 5、6 号泳道为pET28a-pgm重组表达载体的双酶切产物, 电泳图中pgm基因浓度太低, 条带不明显。 7、8 号泳道为pET28a-pgm重组表达载体的PCR 产物, 扩增片段大小在1 700 bp 左右,符合pgm基因长度。 9、10 号泳道作为阳性对照(A17 中pgm基因的PCR 扩增产物)。
将pET28a-pgm重组表达载体的测序结果BLASTx 比对分析, 结果显示重组表达载体中的pgm基因序列与网站中序列完全匹配, 证明重组表达载体中的pgm基因就是EPS 合成过程中编码葡萄糖磷酸变位酶的关键基因。
2.4 目的重组蛋白质的表达与纯化
活化重组成功的大肠杆菌, 用IPTG 诱导其表达目的蛋白质。 诱导后离心菌液,对上清液和沉淀分别进行SDS-PAGE 电泳,结果如图6 所示。 诱导产物用His-tag 亲和层析纯化,结果如图7 所示。目的蛋白质的相对分子质量约为60 000 左右,从电泳图中可以看出,1 泳道的未经诱导对照组在60 000 左右没有蛋白质,IPTG 诱导的上清液和沉淀中60 000 处有条带,说明重组大肠杆菌成功表达了目的蛋白质,并且上清液中的目的蛋白质含量明显多于沉淀,说明蛋白质主要在上清液里。

图6 IPTG 诱导蛋白质表达结果Fig. 6 Results of IPTG-induced protein expression
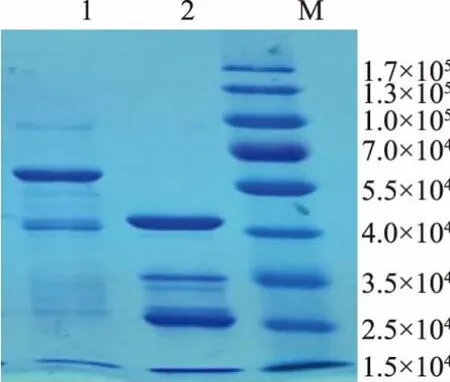
图7 亲和层析纯化结果Fig. 7 Affinity chromatography results
对含目的蛋白质的上清液进行His-tag 亲和层析,纯化蛋白质,层析产物浓缩电泳,1 号泳道中出现目的条带,2 号泳道是层析中出现的杂蛋白质。利用AKTPurifier 分子筛去除杂蛋白质,可得到高纯度(95%)的目的蛋白质。
2.5 目的蛋白质质量浓度的测定
采用最为常见的Bradford 法进行测定,每个样品均3 次重复。 将BSA 作为标准品,该法所测定的标准曲线在质量浓度2.5~15 μg/mL 范围内保持线性关系。 测定目的蛋白质浓度的标准曲线为:y=0.025 1x+0.019 3,浓缩后蛋白质测定的OD 值为:y=0.302(平均值),因此求得x约为11.3 μg/mL。 由于蛋白质是经过稀释100 倍后测定的,故计算得重组蛋白质的最终质量浓度为1.130 mg/mL, 目的蛋白质表达量较高,可实现量产。
3 结 语
因为双歧杆菌在肠道中可以抑制有害细菌,改善胃肠道屏障功能[21],长期以来一直被用作益生菌来改变肠道菌群组成以缓解各种疾病。 最近的研究表明,双歧杆菌可改变树突状细胞的功能,以调节肠道对无害抗原和细菌的免疫稳态,或启动针对病原体的机体保护措施[22-25]。 据报道,很多双歧杆菌会合成EPS,由于EPS 具有与周围环境沟通的能力以及对宿主健康的维护,受到了科学界相当大的关注[26]。一些体外研究表明,特定的EPS 可以在产EPS 的菌中发挥有益的作用[22]。 然而,值得注意的是,由于双歧杆菌EPS 的产率低,这些聚合物作为市售益生元添加剂的用途非常有限[26]。 可通过优化发酵条件例如优化发酵时间、pH、培养温度、碳氮源等或者通过基因工程技术等来提高胞外多糖产量。 在进行基因工程改造菌株时可通过超量表达EPS 合成途径中的基因例如磷酸葡萄糖变位酶(pgm)、UDP-葡萄糖焦磷酸化酶(galU)等来提高EPS 产量,以期解决双歧杆菌作为一种益生菌但其胞外多糖产量低下的问题[26-28]。
EPS 的合成是一个很复杂的过程, 包括糖进入细胞质、前体糖核苷酸的形成以及胞外多糖合成与输出[20]。乳酸菌的EPS 生物合成由eps基因簇调控,决定EPS 重复单元的合成、聚合、输出等过程。在本研究中经过一系列生物信息学分析发现,EPS 的生物合成与一个关键的基因相关,在乳酸菌当中通过过表达该基因使胞外多糖的产量有较大幅度的提升。双歧杆菌eps基因簇没有像LAB“保守”eps基因簇一样的特性,且种间和种内的差异极大[11],本研究中在与其它双歧杆菌eps基因簇进行比较时也发现这一点,说明单一研究某一种双歧杆菌来推断其它双歧杆菌的eps基因簇是不可取的, 只能与其它双歧杆菌进行比较分析。 结合作者所在实验室前期长双歧杆菌A17 全基因组测序结果,成功分析到胞外多糖A17 的合成途径。
在乳酸菌及其亚种当中,提高胞外多糖产量的途径可通过优化发酵条件, 例如改变培养温度、优化发酵时间、碳源等来实现。 为了提高双歧杆菌胞外多糖产量低的问题, 通过选取pET28a 重组表达质粒实现了葡萄糖磷酸变位酶的大量表达,为从分子手段调控双歧杆菌EPS 的产量提供了新的途径。将以往解决胞外多糖产量问题从传统的优化培养条件上升到选取拷贝数与表达效果兼优的质粒,大大缩短了实验时间,更快更高效解决胞外多糖产量低下的问题,对后续开展针对双歧杆菌及其EPS 功能研究以及进一步阐明双歧杆菌EPS 生物合成具有重要意义。
猜你喜欢
中国化肥信息(2019年12期)2020-01-16 08:40:06
西北农林科技大学学报(自然科学版)(2019年8期)2019-07-17 02:43:32
中国化肥信息(2018年7期)2018-08-23 09:12:32
中国化肥信息(2018年6期)2018-08-23 09:11:42
中国化肥信息(2017年7期)2017-12-13 08:46:28
遗传(2015年5期)2015-02-04 03:06:55
海洋科学(2014年12期)2014-12-15 03:35:00
中国药业(2014年19期)2014-05-17 03:12:16
食品科学(2013年23期)2013-03-11 18:30:03
食品科学(2013年17期)2013-03-11 18:26:57
