
碳纳米管活化过硫酸盐降解污染物的研究进展
2023-11-08 17:58解勤兴叶布天于子钧贾彦军
天津工业大学学报 2023年5期
解勤兴,叶布天,于子钧,贾彦军,张 阳,王 骏
(1.天津工业大学省部共建分离膜与膜过程国家重点实验室,天津 300387;2.天津工业大学材料科学与工程学院,天津 300387;3.天津工业大学纺织科学与工程学院,天津 300387;4.天津工业大学环境科学与工程学院,天津 300387)
水是人类赖以生存的重要资源之一。随着我国工业化和城镇化的快速发展,水体有机污染和水资源短缺已成为目前正在面临而且将不断恶化的重大环境问题。近年来,过硫酸盐高级氧化技术(PS-AOPs)在处理难降解有机废水方面取得了较大的研究进展[1-2]。过硫酸盐包括过一硫酸盐(PMS,HSO5-)和过二硫酸盐(PDS,S2O82-),其过氧键(O—O)可以通过电子转移或能量吸收的方式被活化,使O—O 键断裂产生羟基自由基(HO·)或硫酸根自由基(SO4·-)等具有强氧化能力的活性氧物种(ROS),以自由基进攻的方式降解有机污染物。此外,过硫酸盐也可以被一些催化剂活化产生非自由基ROS,例如单线态氧(1O2)和表面复合体(catalyst-PMS*),尽管它们的氧化能力较弱,但是非自由基ROS 在复杂的水质环境(共存离子、天然有机质)中仍可快速、选择性地去除富电子有机污染物,因此,在实际水处理过程中具有独特的优势。
基于碳材料非均相活化过硫酸盐高级氧化技术因其绿色、高效和选择性强引起了人们的广泛关注,是一种具有良好发展前景的新型水处理技术。其优势在于:①碳材料本身无毒性,能避免金属离子浸出带来的二次污染问题;②碳活化过硫酸盐氧化有机物具有较强的选择性,在复杂水体下亦能保持良好的催化活性;③在高浓度卤素离子环境中,能抑制毒性卤副产物的产生。碳纳米管(CNT)是一种具有纳米直径和微米轴长的一维纳米材料,主要由高度石墨化的sp2杂化碳组成,结构简单,表面官能团和缺陷较少,且易于结构改性和调控,常被用作碳基模型材料研究过硫酸盐活化降解有机污染物的作用机理和构-效关系。关于碳纳米管材料的制备、改性和结构调控已有较为详尽的总结,本文主要概述了CNT 材料激活PMS 和PDS 降解有机污染物的性能及其构-效关系,针对CNT 材料循环稳定性不佳的问题,提出了可能的原因和再生方法,重点论述催化机理和ROS 鉴定方法,并对该领域未来的研究方向提出展望。
1 CNT 激活过一硫酸盐(PMS)
2012 年,澳大利亚科廷大学的Wang 课题组[3-6]发现石墨烯(rGO)界面上存在丰富的吸附和催化活性位点,能够激活过硫酸盐产生多种活性氧化物种(ROS),如SO4·-/HO·以及1O2等,可实现包括染料、酚类、抗生素等在内的一系列污染物的快速氧化降解。此后,基于碳基催化活化过硫酸盐降解水中污染物的研究引起了人们的关注,成为当前环境催化领域的研究热点之一[7]。碳基纳米材料根据维度可以分为零维、一维、二维和三维材料,主要包括:零维的富勒烯(C60)和纳米金刚石(ND),一维的碳纳米管(CNT),二维的石墨烯纳米片(GNS),以及三维的有序介孔碳(CMK)、活性碳(AC)和碳气凝胶(CA)等。其中,CNT 激活PMS 的效能和机制如图1 所示。
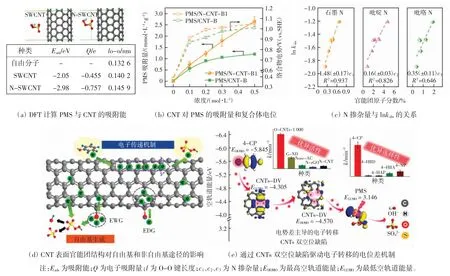
图1 CNT 激活PMS 的效能和机制Fig.1 Performances and mechanisms of PMS activation by CNTs
尽管碳基材料对于有机污染物的吸附能力遵循0维<<1 维<2 维<3 维,但是其激活过硫酸盐降解有机污染物的性能则与材料的结构和活性位点密切相关。例如,sp2杂化的碳纳米管的催化活性要远强于sp3杂化的纳米金刚石和sp2/sp3杂化的富勒烯,此外,氧官能团含量、孔隙率、杂原子掺杂程度和缺陷程度等对碳材料的催化活性有较大影响[2]。
1.1 杂原子掺杂
Sun 等[8]使用单壁碳纳米管(SWCNT)激活PMS 降解苯酚(20 mg/L),发现在150 min 内苯酚的去除率达到76%,将SWCNT 与硝酸铵混合热处理得到N 掺杂碳纳米管(N-CNT),其催化性能显著提高,在45 min内即能将苯酚完全去除。Duan 等[9]将三聚氰胺和SWCNT 混合在700 ℃下热处理制备得到NoCNT-700,可以在20 min 内降解100%的苯酚,通过密度泛函理论(DFT)计算发现,N 掺杂能够提高PMS 的吸附能[10],加快电子转移速率并使PMS 的O—O 键增长,从而更容易被吸附在CNT 表面生成ROS 降解污染物(图1(a))。Ren 等[11]用实验证实了N 掺杂能够显著提高N-CNT 对PMS 的吸附量以及N-CNT-PMS*复合体电位,从而加速非自由基电子转移过程,实现苯酚的快速氧化(图1(b))。氮含量与反应速率常数对数(ln kobs)的相关性分析显示,N-CNT 的催化活性与N含量尤其是石墨型N 含量呈显著的正相关性(R2=0.937)(图1(c)),说明石墨型N 原子是N-CNT 上的主要活性位点,这与其他研究得到的结论相一致[5,12]。
除了N 掺杂以外,B、S 等元素掺杂也能提升CNT的催化活性。Li 等[13]通过混合硼酸和CNT 在800°C 氮气保护下进行热处理,得到了B 元素含量为0.61%的B 掺杂MWCNT,可以在60 min 内降解99.4%苯酚(10 mg/L),同时还表现出pH 值适用范围广(1~9)的优点,这是因为硼原子可以改变相邻碳原子的电荷密度并增加缺陷边缘,从而提高催化活性。相比单元素掺杂,多元素共掺杂能进一步提高CNT 激活PMS 的催化性能。Liu 等[14]通过将CNT 和硫脲混合热处理制备得到N、S 共掺杂的CNT,在30 min 内对二苯甲酮-4(BP-4,10 mg/L)的去除率达到100%,反应速率比氮原子单独掺杂的N-CNT 高5 倍,这是由于N、S 共掺杂产生协同效应,可以重新分配自旋电荷密度,并促进电子转移[15]。
1.2 含氧官能团调控
CNT 表面的含氧官能团对PMS 催化性能影响较大,但其作用仍然存在争议。Liu 等[14]比较了羧基化CNT、羟基化CNT 与原始CNT 活化PMS 降解BP-4的性能,发现三者在60 min 内对BP-4 的去除率分别为82%、26%和35%,这说明CNT 上的羧基能够促进PMS 活化,而羟基则具有抑制作用。也有研究发现,首次反应后的CNT 表面羧基相对含量显著提高(7.6% ~16.2%),重复使用后对苯酚的去除率却由55.3%下降到32.8%,羧基增加反而不利于PMS 的活化[16]。最近,Han 等[17]通过强酸处理将CNT 氧化得到OCNT,再在800 ℃下退火得到还原的OCNT800。尽管OCNT 的比表面积、表面缺陷程度和羰基含量均高于CNT 和OCNT800,但是却没有催化活性;而去除了大部分含氧官能团的OCNT800 反而能够在60 min 内降解98%以上的苯酚。这是由于过多的含氧官能团会破坏CNT 的sp2杂化结构,使导电性下降,从而抑制电子转移过程降解污染物。将CNT 上的含氧官能团分为供电子基团(EDG,如羟基、酯基)和受电子基团(EWG,如羧基、羰基),如图1(d)所示,受电子基团能够直接激活PMS 产生活性自由基,供电子基团可以提高碳层表面的电子密度和PMS 吸附量,从而促进非自由基电子转移途径降解污染物。这些研究说明碳材料表面官能团的种类、含量与其电子密度和催化活性密切相关。
1.3 缺陷程度调控
此外,官能团调控的过程往往会影响碳纳米管表面的缺陷程度。例如,Shao 等[18]将混酸处理后的o-CNT在不同温度下(600~1 100 ℃)煅烧,发现当温度从600升高到1 000 ℃时,o-CNT 的Raman 光谱ID/IG值由1.04 上升到1.37,进一步提高到1 100 ℃后ID/IG值降至1.18。o-CNT-1000 在激活PMS 降解对氯苯酚(4-CP)的实验中展现出最高的催化活性,4-CP 的降解速率与碳纳米管的ID/IG呈显著的正相关。DFT 计算发现(图1(e)),双空位缺陷的能垒较低,电子转移比单空位缺陷和Stone-Wale 缺陷更加容易,推测双空位缺陷可能是PMS 催化的高能活性位点。然而缺陷过多也会导致CNT 上的sp2杂化碳原子转化为sp3杂化结构,破坏CNT 上原始的π 共轭结构,不利于电子转移反应的进行[19]。目前关于碳催化的研究往往难以区分出官能团和缺陷在催化中的贡献,而且关于缺陷的定量手段比较单一,未来的研究应致力于碳纳米管缺陷工程调控,为新型环境功能材料的开发提供技术支持。
1.4 与金属复合
过渡金属如Fe、Co、Ni、Mn 及其氧化物已被证明是高效的PMS 活化剂[20],能够通过自由基或者非自由基途径降解有机污染物。将其与碳纳米管复合,不仅可以提高金属纳米材料的分散性,还能够利用CNT 对有机污染物超强的吸附能力以及金属材料良好的电导性,产生显著的协同效应高效降解水中污染物。
CNT 与金属纳米材料复合的方法主要包括热裂解法和扩散-沉积法。前者是用三聚氰胺或双氰胺等与金属盐混合热处理,可同时实现杂原子掺杂和金属负载,适用于合成Co、Ni、Fe 等过渡金属/氧化物催化剂,其结构和性质还可以通过改变热处理温度、复合比例等来调控。后者是将碳纳米管浸渍到含有金属前驱体的溶液中,金属前驱体扩散到碳纳米管表面和管内,然后通过蒸发、水热、电化学等方法使金属前驱体沉积,最后在惰性气氛中退火或用氢气还原使前驱体转化为纳米颗粒。各种CNT 复合催化剂的制备方法和结构如图2 所示。
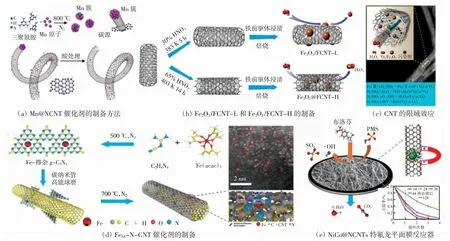
图2 各种CNT 复合催化剂的制备方法和结构Fig.2 Preparation methods and structures of various CNT hybrid catalysts
Kang 等[21]以三聚氰胺和氯化锰作为前驱体,如图2(a)所示,在N2保护下800 ℃高温热处理制得Mn@NCNT催化剂,在NCNT 内部形成了稳定的锰单质和碳化锰。该材料能够高效活化PMS 产生活性自由基,对微塑料具有优异的降解性能。同样地,Yao 等[22]用热裂解法制备了NCNT 包裹的不同金属催化剂(Co、Fe、Ni),其中,Co@N-C/PMS 体系具有高氧化能力,在120 min内即可实现20 mg/L 酸性橙(AO7)的完全降解,催化活性的大小顺序为Co@N-C >Fe@N-C >Ni@N-C。值得注意的是,在碳纳米管内层或外层分布的金属纳米材料在催化中所起的作用可能会不一样。Yang 等[23]对CNT 进行了不同的酸处理,并采用浸渍-沉积法制备了表面负载或内层包裹Fe2O3的CNT,如图2(b)所示。当Fe2O3纳米颗粒主要分布在CNT 外层表面时,该材料在催化Fenton 反应中产生的主要活性物质为HO·自由基。而当Fe2O3被包裹在CNT 内部时,单线态氧(1O2)则是主要的活性物质(图2(c)),不仅具有较高的选择性氧化能力,而且大大降低了金属离子的释放,这说明碳纳米管的限域效应能够对催化反应产生显著影响,关于限域催化的研究亟待深入开展[24]。
新兴的碳基单原子催化剂(SACs)因其环境友好、性能超高以及活性金属位点的最大利用率而成为环境催化中极具吸引力的先进材料。Qian 等[25]通过将Fe掺杂g-C3N4与CNT 球磨和烧结,构建了以CNT 为基底的Fe 单原子催化剂(FeSA-N-CNT),如图2(d)所示。Fe 单原子以FeN4的形式存在,能够在较广的pH值范围(3.0~9.0)内高效激活PMS 产生FeN4——O 氧化双酚A,FeSA-N-CNT/PMS 体系的反应速率(k 值)高达256 μmol/(s·g),比之前报道的非均相类芬顿反应的速率高7.1~2 844.4 倍。DFT 计算表明,CNT 基底能够优化Fe 3d 杂化轨道,并促进PMS 在FeN4位点上的吸附,从而大大增强FeN4——O 的氧化活性。
此外,碳纳米管及其复合材料,因其纳米尺寸使得它们在实际环境中难以回收利用和管理控制而受到应用限制。为了解决这个问题,Kang 等[26]合成了NiCo@NCNTs 并将其抽滤涂覆在特氟龙滤膜上形成平面膜反应器,如图2(e)所示,能够高效激活PMS 降解布洛芬,反应速率高达0.31 min-1。Shen 等[27]通过将碳纳米管与石墨烯共组装形成三维宏观体,不仅能够保持其纳米基元的优异性能,而且能够结合三维结构孔隙率高、比表面积大以及宏观块体易于回收等优势,在环境领域展示出极具吸引力的前景。因此,开发纳米器件材料并将其用于过硫酸盐活化降解污染物值得深入研究。
2 CNT 激活过二硫酸盐(PDS)
PDS 是一种白色固体粉末,具有比PMS 更高的标准还原电势(E0=2.01 V vs 1.75 V)和更低的过氧化物键能(140 kJ/mol vs 140~213.3 kJ/mol),因此,理论上PDS 比PMS 更易于被催化剂活化且对酚类物质具有更高的氧化能力[11]。
Guan 等[28]用CNT 分别激活PMS 和PDS 降解苯酚(20 mg/L),在90 min 内对苯酚的去除率分别为62%和100%[8]。同样地,CNT/PDS 体系完全去除2-溴酚(10 μmol/L)仅需40 min,相比之下CNT/PMS 体系则需要60 min 才能完全降解。通过研究发现,当CNT对PMS 和PDS 的摩尔吸附量相同时,CNT/PDS 体系的氧化还原电位要明显高于CNT/PMS 体系,因此氧化能力更强[11]。为了深入探讨CNT/PDS 体系的选择性氧化机制,Ren 等[29]测试了12 种酚类化合物的半波电位(φ1/2),并将其与CNT/PDS 体系中的动力学速率常数(lnkobs)进行定量构效关系(QSAR)分析,结果如图3(a)所示。酚类化合物的φ1/2越高,反应速率越低,两者之间呈良好的线性负相关性。电化学分析表明,PDS首先在CNT 表面活化形成具有氧化能力的CNT-PDS*复合体,使氧化还原电势抬升,该复合体能够通过夺取电子的方式选择性地氧化CNT 表面共吸附的酚类化合物(图3(b))。其选择性主要依赖于CNT-PDS*复合体的氧化电位(Ecom) 和酚类化合物的氧化电位(Eorg),如图3(c)所示。当Ecom>Eorg时复合体将从有机物中提取电子将其氧化;当Ecom≤Eorg时有机物不能向复合体转移电子,因此,难以被氧化。
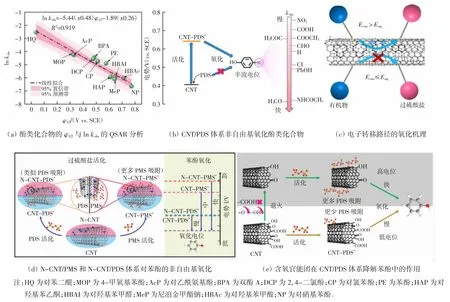
图3 CNT/PDS 体系的非自由基氧化过程Fig.3 Non-radical oxidation process of CNT/PDS system
2.1 N 掺杂
尽管PDS 比PMS 的标准还原电势更高,但由于PDS 是对称结构,PMS 是非对称结构,因此,材料表面改性可能有利于PMS 催化,而对PDS 催化的影响较小。在上文中提到,N 掺杂能够提升CNT 激活PMS 降解苯酚的性能,但是同样的改性却对PDS 氧化没有促进效果[8]。如图3(d)所示[11],表面N 掺杂可以显著提高N-CNT 对PMS 的吸附量及其复合体电位,从而加速电子转移过程,促进有机物的氧化。但是,N 掺杂并没有提高CNT 对PDS 的吸附,与CNT/PDS 体系相比,N-CNT/PDS 体系的氧化电势没有明显变化,因此,催化性能基本保持不变。此外,该研究还揭示了苯酚的氧化速率与N-CNT-PDS*复合体电势有良好的线性相关性,说明N-CNT/PDS 体系也是一个基于电子转移机制的氧化体系[11]。
2.2 含氧官能团和缺陷程度调控
为了探究CNT 上含氧官能团在PDS 活化中的作用,Ren 等[30]通过高温退火获得了一系列表面含氧量不同的CNT,并激活PDS 降解苯酚。在高温退火后,CNT/PDS 体系对苯酚的氧化速率显著提高,反应速率常数(lnkobs)和含氧量之间呈现良好的线性负相关,这说明较低的氧含量(原子分数<5%)能够强化电子转移过程,而氧含量过高(原子分数>8%)不仅会降低CNT 的导电性,而且会占据边缘位点并通过空间位阻效应阻止PDS 与过硫酸盐的相互作用,不利于电子转移氧化苯酚。与PMS 催化不同的是,CNT 上羰基和羧基含量与lnkobs之间呈现显著的负相关性,说明它们并不是激活PDS 的活性位点,而羟基的相关性相对较低(R2=0.762),说明羟基的影响并不明显。QSAR 分析显示,lnkobs与CNT 的缺陷程度(ID/IG)相关性较差(R2=0.037),说明缺陷位点不是CNT 激活PDS 的主要活性位点。CNT 的表面含氧量与其Zeta 电位密切相关,含氧量降低会使CNT 在中性溶液中的Zeta 电位变得更正,促进PDS 的吸附,在CNT 表面形成更多的CNT-PDS*复合物,从而加速电子转移过程降解苯酚(图3(e))[30]。Wang 等[31]通过带有氨基基团的高分子聚合物聚乙烯亚胺(PEI)非共价改性CNT,增强CNT 对PDS 的吸附,改性后的CNT 能在3 min 内完全降解双酚A(20 mg/L),催化活性可达到原始碳材料的400倍,这说明通过改性增加PDS 的吸附量能够显著加速催化反应。
2.3 金属负载CNT 催化剂
过渡金属及其氧化物在PDS 催化中具有良好的性能,多种金属负载CNT 催化剂被开发出来用于高效激活PDS 降解污染物,其催化活性和反应路径与金属种类及负载位置相关,如表1 所示。

表1 不同CNT 催化剂活化过硫酸盐的性能及其作用机理Tab.1 Performances and mechanisms of persulfate activation by various CNT catalysts
Yao 等[22]研究了不同金属(Fe、Co、Ni)负载CNT 催化剂激活PDS 降解酸性橙(20 mg/L)的性能,在120 min内,Fe@N-C、Co@N-C 和Ni@N-C 分别能够去除35%、60%和80%的酸性橙。Jiang 等[46]通过将纳米Co3O4封装在N 掺杂碳纳米管(Co3O4@NCNTs)中,表现出优异的PDS 活化性能,在30 min 内即能实现橙黄G(0.2 mmol/L)的完全去除,催化剂异质界面处的Co-N 位点具有高反应活性,能够通过1O2和电子转移过程高效降解污染物。Liu 等[47]通过化学气相沉积(CVD)法分别制备出CNT 表面负载Fe3O4(Fe3O4/CNT) 和内部封装Fe3O4(Fe3O4@CNT)催化剂,并激活PDS 降解四环素(8 mg/L)。Fe3O4@CNT 表现出更为优异的催化效果,其kobs是Fe3O4/CNT 的6.3 倍,这是由于产生了有较高选择性氧化能力的单线态氧。此外,DFT 计算还表明限域效应使得Fe3O4@ CNT比Fe3O4/CNT 有更高的费米能级(-0.214 2 eV vs-0.208 9 eV),有利于电子转移过程降解污染物。这说明CNT 的限域效应能够对催化性能和反应路径产生重要影响,其反应原理和构-效关系值得深入研究。
3 CNT 催化的影响因素
在实际环境水体中,水质条件较为复杂,pH 值和温度差异较大,且水中存在较多的无机阴离子和天然有机质(NOM),可能会明显影响过硫酸盐的活化过程。 部分环境因素及CNT 结构对催化的影响如图4 所示。

图4 环境因素及CNT 结构对催化的影响Fig.4 Effects of environmental conditions and CNT structures on catalysts
3.1 pH 值
Ren 等[11]研究发现,N-CNT 对PMS 的吸附能力随pH 值的升高而降低,对苯酚的催化能力也随之降低。这是由于在高pH 值条件下N-CNT 表面电负性增强,与HSO5-和污染物之间的静电斥力变大,不利于CNTPMS*复合物的形成,从而阻碍N-CNT 通过非自由基途径降解苯酚。
3.2 表面Zeta 电位
PMS 和PDS 的吸附量均与N-CNT 的表面Zeta 电位成线性正相关,如图4(a)所示,即Zeta 电位越正,过硫酸盐的吸附量越多,产生的复合体电势越高,苯酚氧化也越快[11]。
3.3 温度
Duan 等[48]研究了温度对N-CNT/PMS 体系降解硝基苯性能的影响。在低温范围(5~45°C)内,N-CNT/PMS 体系通过非自由基途径降解硝基苯,反应速率随温度上升而提高,反应活化能为26.1 kJ/mol。在高温区间(45~75°C),硝基苯的降解速率显著加快,展示出热活化和碳活化的协同效应,反应活化能降低到13.2 kJ/mol,淬灭实验表明高温热驱动将N-CNT/PMS 体系变成以自由基途径为主导的反应过程(图4(b))。
3.4 阴离子
在生活或工业污水中通常含有大量的无机阴离子,SO42-、H2PO4-和NO3-等离子可以通过螯合反应吸附在金属或非金属催化剂表面,从而竞争性地阻碍过硫酸盐和催化剂之间反应性复合物的形成[11,49]。HCO3-对SO4·-和HO·有清除效果,所以会抑制以自由基途径为主的反应[28],过量的HCO3-也会影响溶液pH 值并干扰过硫酸盐催化反应[35]。此外,Cl·离子能够与SO4·-反应生成活性较低的HClO-和Cl·,因此,不利于污染物降解[35],但Chen 等[50]在研究CNT 活化PMS 降解AO7 时发现,高Cl-浓度下AO7 降解率会提高,这是由于HClO-对AO7 等偶氮类染料有高反应活性,所以具有明显的脱色效果。值得一提的是,SO4·-或HO·自由基能将Br-离子氧化为致癌物溴酸根(BrO3-),而非自由基途径则不能将其氧化,而且对于天然有机质以及复杂的水质条件,非自由基途径能够选择性氧化降解富电子有机污染物,因此,在实际环境中具有更好的应用前景。
3.5 碳纳米管结构
除了环境因素以外,碳纳米管的层数、表面结构以及催化剂负载方式也会显著影响碳纳米管的催化性能和机理(图4(c))。尽管单层碳纳米管(SWCNT)的比表面积大于双层(DWCNT)和多层碳纳米管(MWCNT),但是SWCNT 相对较高的表面能使其在溶液中易于缠绕和团聚,使暴露于反应环境的活性位点减少,进而降低催化活性[13]。碳纳米管表面的缺陷结构可以促进PMS 的活化但对PDS 的影响较小,其中双空位缺陷可以增加CNT 对PMS 的吸附[18],但过多的缺陷也会降低其导电性,不利于电子转移反应的进行[19]。碳纳米管表面的氧官能团含量会明显影响碳纳米管的Zeta 电位和导电性,过高的氧含量会使CNT 表面呈负电性从而减少对过硫酸盐的吸附,导致催化活性下降[30]。如前所述,CNT 表面负载金属纳米颗粒可以有效提高其催化活性。另外,当金属负载于CNT 内部时会存在限域催化效应,可以增强选择性氧化能力,而且降低了金属离子的释放[24]。当金属催化剂以单原子的形式(M-N4)负载于CNT 表面时,可以促进PMS 或PDS 在M-N4位点上的吸附[25],M-N4结构也可以通过改变电子分布有效降低电子转移阻力,从而促进电子转移[46]。
4 CNT 催化剂的循环再生
碳纳米管催化剂的稳定性和重复利用性是决定其能否实际应用的关键因素。一般来说,碳纳米管材料的催化性能随反应次数的增加逐渐衰退,但是一些金属负载CNT 催化剂仍然表现出了优良的循环性能。例如,Yao 等[22]通过热裂解制备出Co@N-CNT 催化剂,首次活化PMS 可以在90 min 内降解99%的酸性橙,经过5 次循环运行后,依然可以在180 min 内实现约98%的酸性橙去除,催化性能仅有限下降。
CNT 材料催化性能衰退的机理和再生方法如图5所示。
(5) 维修性.在桥架完全开启时,悬挂油缸通过液压控制回路即可卸载,此时即可对悬挂机构各组成零部件进行维修和更换.考虑到悬挂方案维修主要是对损坏零部件进行更换,故可用机械平均更换时间TB,单位min,评价此悬挂方案的维修方便性,即

图5 CNT 材料催化性能衰退的机理和再生方法Fig.5 Deactivation mechanisms of CNT materials and regeneration methods
催化剂循环性能下降主要有以下3 个原因:
(1)中间产物覆盖。反应过程中,污染物氧化产生的中间产物不能被完全矿化,残留的中间产物会吸附在催化剂表面,导致活性位点被覆盖,难以发挥作用[39,45,51]。
(2)表面氧化。碳纳米管被生成的ROS 氧化,表面含氧量提高且电负性增强,活性位点如羰基和石墨型N 原子等含量减少,阻碍了过硫酸盐的吸附和催化。
(3)结构坍塌和失活。自由基的强氧化性能使得碳纳米管的结构坍塌,导致其比表面积和孔含量减少,对于负载金属的催化剂还存在活性组分流失的情况[39]。
为了判断催化剂在循环过程中的失活机理并将其再生,通常采用有机溶剂(乙醇、正己烷和丙酮等)清洗的方法,将吸附在固体表面的污染物及其中间产物洗脱,或采用紫外辐照的方式,活化材料表面残留的过硫酸盐深度降解中间产物。如果催化剂的活性完全恢复,说明其失活机理主要是中间产物覆盖。如果催化剂的活性未恢复或仅部分恢复,可以在惰性气体保护下进行高温处理(>350°C)或通过化学还原的方式,去除材料表面的含氧官能团并恢复其sp2杂化共轭结构。若催化剂的活性完全恢复,说明其失活机理主要是表面氧化,若未恢复或仅部分恢复,则可能是结构坍塌和失活,需要进一步通过结构表征来进行验证。Kang 等[39]通过测量使用前后Ni@NCNT 的比表面积发现,催化剂比表面积由155 m2/g 降低到54 m2/g,说明碳纳米管结构破坏是其失活的重要原因。Hou等[16]对比使用前后的N-CNT 的表面元素含量发现,N原子分数由2.56%下降到2.38%,吡啶型、吡咯型和石墨型氮原子的含量均有所下降,而氧化氮的相对含量却由8.7%上升到14.2%,说明性能下降主要与氮掺杂位点的流失和氧化有关。此外,还可以通过扫描电子显微镜(SEM)、X 射线衍射(XRD)和拉曼光谱(Raman)等进一步分析材料在反应前后的结构变化及其失活机理,为今后开发绿色高效且便于循环的非均相催化材料提供理论依据和技术支撑[16,42]。
5 CNT 催化机理及其鉴别方法
碳纳米管催化剂活化过硫酸盐降解污染物的机理主要有2 种:第1 种是自由基降解途径,基于活化产生强氧化性的自由基(HO·和SO4·-)降解有机污染物;第2 种是非自由基降解途径,即在反应过程中不依赖自由基,通过1O2或表面电子转移过程对富电子物质进行选择性(例如,染料、苯酚、氯酚和大分子抗生素)亲电攻击。2 种不同的反应机制引起了人们的广泛讨论。
5.1 自由基机理
5.1.1 自由基的产生与反应
CNT 通过向PMS 或PDS 转移电子使过硫酸盐中O—O 键断裂,以产生SO4·-和HO·。转移的电子按来源可分为:①来自碳催化剂缺陷(例如空位和锯齿形/扶手椅边缘)的离域π 电子[4];②路易斯碱性位点的孤对电子,如C—O 基团中的O 原子[52];③CNT 包裹的金属氧化物通过协同作用转移到sp2杂化碳晶格的自由流动π 电子[22]。SO4·-和HO·可以通过3 种机制氧化有机物:①从饱和化合物(例如烷烃)中夺氢;②电子提取;③与不饱和芳香族化合物发生亲电加成或取代反应。SO4·-和HO·作为具有强氧化性的ROS,可以与大多数有机物发生反应。相比之下,SO4·-表现出比HO·更强的电子提取能力,有利于单电子氧化具有活化基团的芳香族化合物(例如苯甲醚、对羟基苯甲酸等)[53],而具有钝化基团的芳香族化合物(例如硝基苯和对硝基苯甲酸等)则几乎不能与SO4·-反应[54]。
5.1.2 自由基的鉴定
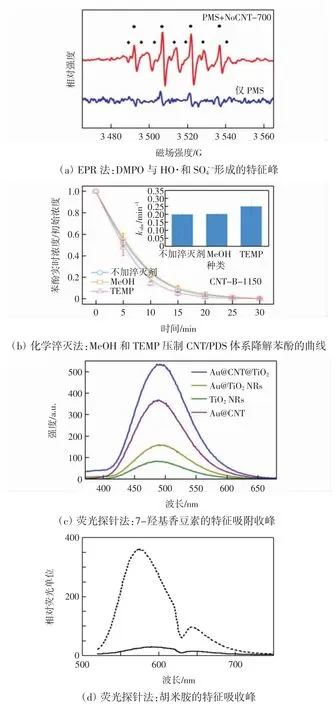
图6 不同活性氧物质检测方法的结果示意Fig.6 Results diagran of detection methods for different ROS
(1)电子顺磁共振(EPR):自由基半衰期短,难以直接通过EPR 检测,需要用自旋捕获剂(如5,5-二甲基-1-吡咯啉N-氧化物,DMPO)将短寿命的自由基转化为EPR 可测量的长寿命顺磁性自旋加合物[9,55],通过峰的形状和超精细分裂常数(αN、αβ-H)来识别相应的自由基。如图6(a)所示,DMPO-HO·(1 ∶2 ∶2 ∶1,αN=αβ-H=14.9 G)和DMPO-SO4·-(1 ∶1 ∶1 ∶1 ∶1 ∶1,αN=13.51 G,αβ-H=9.93 G)分别为DMPO 与HO·和SO4·-形成的特征峰,说明NoCNT-700/PMS 体系产生了以上2种活性自由基[9]。DMPO 也可以用来探测O2·-是否存在,但需要在甲醇中进行测试,因为在水溶液中形成的DMPO-O2·-(αN=14.3 G,αβ-H=11.2 G)不稳定,而且信号也容易受到HO·和SO4·-干扰。
(2)化学淬灭:化学淬灭是识别ROS 的常用策略,添加不同的ROS 淬灭剂到反应体系中,通过检测抑制程度来评估各种ROS 在反应过程中的相对贡献。也就是说,抑制程度越高,该ROS 的贡献就越大。表2给出了甲醇(MeOH)、叔丁醇(TBA)和氯仿(CF)等淬灭剂与不同自由基的反应速率。

表2 各种淬灭剂和不同活性氧物质的反应速率Tab.2 Reaction rates of various scavengers toward different ROS
由表2 可以看出,MeOH、乙醇(EtOH)和异丙醇(IPA)等能够高效淬灭HO·和SO4·-,TBA 与HO·的反应速率远高于SO4·-,常被用来区分2 种自由基的相对贡献,而CF 和对苯醌(p-BQ)则能选择性淬灭O2·-。加入MeOH(MeOH/PDS 摩尔比为500)对苯酚降解没有抑制能力,如图6(b)所示,说明CNT-B-1150/PDS 是一个以非自由基过程为主导的催化体系[30]。应该强调的是,MeOH 等淬灭剂与碳材料之间的作用力较弱,很难淬灭固体表面的自由基。通常采用硝基苯(NB)、苯甲酸(BA)和邻苯二甲酸二乙酯(DEP)等含苯环的物质作为淬灭剂,或将其作为目标污染物。若MeOH 不能抑制该体系的降解效果,但是该体系又能够降解NB和BA 等污染物,说明该催化体系存在固体表面自由基[29]。其中,NB 由于对HO·的反应速率远高于SO4·-,也可以区分固体表面自由基当中HO·和SO4·-的相对含量。此外,苯酚和KI 也常被用作表面自由基淬灭剂[29],但是苯酚因其富电子特性,可以被非自由基电子转移过程降解,而KI 会与PMS 或PDS 发生反应,造成氧化剂的消耗,从而阻碍污染物的降解,这些都会导致催化机理的误判。因此,在使用淬灭剂时应根据反应体系进行筛选,避免对反应过程产生较大影响。同时对反应机理的判断不能只基于淬灭实验,需要结合其他技术手段。
(3)荧光探针法:除了上述方法外,还可以使用化学探针与自由基反应产生的副产物或加合物来进行定性或半定量分析。如图6(c)所示,香豆素可以被HO·定量转化为有荧光性的7-羟基香豆素,在波长为474 nm 处具有最大吸收峰[63],其峰强度能够反映出HO·的含量[64]。对苯二甲酸(TPA)是另一种用于光致发光的化学探针,HO·可以将无荧光的TPA 定量转化为高荧光的2-羟基对苯二甲酸(HTPA),最大吸收波长为425 nm[65]。其他的HO·荧光探针还包括对氯苯甲酸(p-CBA)和水杨酸等[57]。SO4·-也可以与香豆素反应,通过检测产物7-羟基香豆素可以间接确定SO4·-含量[66]。此外,二甲基亚砜(DMSO)也可以检测SO4·-,每2 mol自由基与DMSO 反应生成1 mol 甲醛(HCHO),通过液相色谱进行半定量分析[67]。由于氯化硝基四氮唑蓝(NBT)能被O2·-还原为蓝色的单甲臢,最大吸收峰在530 nm 处,因此,O2·-可以用NBT 显色法进行定性分析,但是该方法的灵敏度较低[68]。如图6(d)所示,O2·-可以将氢化乙啡啶(HE)氧化为荧光产物胡米胺(E+,λEx=520 nm,λEm=610 nm)[69],从而能够被半定量分析,常被用作细胞内O2·-的检测[70]。
5.2 非自由基机理
5.2.1 单线态氧及其鉴别方法
1O2是一种非自由基的ROS,具有中等的氧化电位(E0=1.52 V)和底物特异性,对含有不饱和C——C 键以及硫化物或胺基等富电子化合物具有高氧化性能,这种天然性质赋予了1O2选择性降解药物污染物的能力[57,71]。产生1O2的方法有多种,其中光催化是广泛使用的方法,染料、卟啉、过渡金属配合物和半导体在内的光敏剂可以充当介质,将光能传递给氧分子形成1O2,已经广泛应用于光解污染物和光动力治疗[72-74]。对于碳基催化剂,其表面的离域π 电子(C-π)可以激活PMS 产生高硫酸根自由基(SO5·-),然后SO5·-自重组产生1O2,如式(1)—式(2)所示[71]。
不仅如此,CNT 的多种结构都可以激活PMS 或PDS 产生1O2,包括CNT 表面羰基(C——O)、CNT 包裹过渡金属氧化物以及单原子金属位点(M-N4)等。当PDS 与CNT 络合时,CNT 表面羰基能够水解PDS 生成超氧自由基,与水反应后转化为1O2并产生过氧化氢[61],如式(3)—式(4)所示,整个反应的化学计量预测消耗3 mol 过硫酸盐产生1 mol1O2。
如前文所述,CNT 内部包裹的过渡金属氧化物会触发限域效应,过硫酸盐分子在管内与活性位点作用产生大量的1O2[47]。单原子金属位点(M-N4)也能与CNT 之间产生协同效应,提供电子激活PDS 生成1O2[46]。综上所述,不同催化途径产生的1O2中氧原子的来源主要包括溶液中的溶解氧、过硫酸盐分子氧、高硫酸根自由基和超氧阴离子等,但是其具体反应途径及氧化性能值得进一步研究。
单线态氧可以通过EPR、化学探针法、溶剂交换法以及化学淬灭法等来鉴定。2,2,6,6-四甲基哌啶(TEMP)是一种常用的1O2自旋电子捕获剂,能够与1O2形成稳定的加合物(2,2,6,6-四甲基哌啶-1 氧自由基,TEMPO),在EPR 光谱中出现强度比为1 ∶1 ∶1 的3 重峰(图7(a))[75]。但是这并不能完全证实反应中有1O2生成,因为TEMP 作为杂环胺也可以通过一电子提取形成TEMP 自由基,与O2反应生成TEMPO。因此,常采用二苯基蒽(DPA)作为化学探针,它能够与单线态氧反应形成稳定的DPA 内过氧化物(DPAO2),在LC-MS 上出现明显的特征峰,如图7(b)所示[23]。此外,由于1O2在重水(D2O)中的寿命(20~32 μs)远长于在水中的寿命(<2 μs),通过将反应溶液由H2O 置换为D2O,若有机物的氧化速率明显加快,说明反应中有1O2生成并起到重要作用。

图7 非自由基活性物质的不同检测方法结果示意Fig.7 Results diagram of different detection methods for non-radical ROS
如表2 所示,1O2作为一种非自由基ROS,与许多化学淬灭剂如FFA、NaN3、L-组氨酸等具有较高的反应速率,因此,可以采用化学淬灭法进一步判定1O2在催化过程中的作用。图6(b)给出了TEMP 作为1O2淬灭剂压制CNT/PDS 体系降解苯酚的曲线,TEMP 的加入对苯酚降解无抑制作用,说明1O2在苯酚降解中不起主要作用。需要说明的是,TEMP 是一种碱性物质,加入溶液后会使溶液pH 值向碱性缓冲,从而抑制过硫酸盐在CNT 上的吸附并可能导致碱活化。因此,在选用TEMP 作为淬灭剂时,需要将加入TEMP 后的溶液pH 值调节到中性或酸性范围,以保证实验结果的可靠性。L-组氨酸和NaN3常被用于1O2捕获,但是Yun 等[76]研究发现,在没有催化剂存在的情况下,这2种捕获剂都能直接与PMS 发生反应,造成氧化剂的消耗,使1O2在反应中的作用被高估。另外,当FFA 作为1O2捕获剂时,由于它的氧化电势较低,CNT/PDS 体系能够直接通过电子转移途径将其降解,这也会使底物降解受到抑制,这说明FFA 淬灭不适用于以非自由基电子转移为主的氧化体系[29]。
5.2.2 电子转移机制及其鉴别方法
电子转移机制是PMS/PDS 吸附在CNT 表面形成复合体,使CNT 表面的氧化电势(Ecom)提升,污染物通过π-π 作用和疏水作用等吸附到CNT 表面,当污染物的氧化电势(Eorg)低于复合体的氧化电势(Ecom)时,污染物将电子通过CNT 上的sp2杂化结构转移到过硫酸盐复合体,从而失电子被氧化[17]。PMS 和PDS 均能和CNT 反应产生电子转移过程,但是由于分子结构和性质不同,在活性位点和催化性能上表现出明显的差异。CNT 上的氮掺杂、双空位缺陷和供电子基团等都可以增加对PMS 的吸附,形成更多的CNT-PMS*复合体[11],使氧化电势升高,加速催化反应,但是这些位点对PDS 催化的促进作用并不明显。对于氧官能团,受电子基团羧基(—COOH)和羰基(C——O)会降低Zeta电位而不利于PDS/PMS 吸附,供电子基团羟基(—OH)可以提高碳层表面的电子密度和PMS/PDS 吸附量,从而促进电子转移,但过高的氧含量会破坏CNT 的sp2结构,不利于电子转移的发生[30]。此外,CNT 包裹过渡金属氧化物可以通过协同效应增加CNT 表面的正电性,加强对PMS 和PDS 的吸附,而且M-C 结构可以增加电子传导效率[47],单原子催化剂的M-N4结构也可以通过改变电子分布有效降低电子转移阻力,从而促进电子转移过程[46]。
电子转移机制的鉴别方法主要包括电化学法、原位拉曼法以及选择性氧化法等。
(1)电化学法。当通过化学淬灭法、EPR 法和荧光探针法等分析了其他ROS 的作用后,可以通过线性伏安曲线(LSV)或开路电位来判定该氧化体系中是否有电子转移机制存在。图7(c)给出了典型的LSV 图像,在一个三电极体系中,将CNT 负载在工作电极表面,当溶液中单独加入PDS 或苯酚时,电流只有微弱增加,而当PDS 和苯酚同时加入后,电流密度急剧增加,说明该过程中存在电子转移作用[77]。开路电位能够反映催化剂电极表面过硫酸盐活化以及污染物氧化过程中的电荷转移动力学,如图7(d)所示。在加入PDS后,负载碳催化剂的电极电位急剧上升,逐步达到一个平台期,即复合体电位。当复合体电位高于有机污染物的氧化电位时,将会发生电子转移,此时在体系中加入有机物,开路电位会发生下降,表面复合体将分解为硫酸根离子。对于未负载催化剂的玻碳(GCE)电极,PDS 加入后GCE 的电极电势只有略微上升,因此,不能通过电子转移降解污染物[30]。
(2)原位拉曼法。拉曼光谱对水具有较强的抗干扰能力,因此,可以通过原位Raman 实时监测CNT-过硫酸盐活性复合体的形成和演化,如图7(e)所示[29]。将PDS 溶液加入到CNT 上以后,除了新增S2O82-在810 cm-1附近的峰外,在796 cm-1左右还出现了一个新的峰,该峰对应于亚稳态CNT-PDS*中间体中过氧键(O—O)的弯曲振动[78]。当苯酚加入后,S2O82-的峰强度变弱,同时CNT-PDS*对应的峰消失,而在990 cm-1附近出现了一个SO42-的特征峰,这说明活性复合体将苯酚氧化后分解生成硫酸根离子。
(3)选择性氧化法。选择性氧化法是通过考察催化剂对一系列酚类化合物的降解效果来判断是否存在电子转移的方法。对于一些带有受电子基团(羧基、硝基)的有机物如苯甲酸、硝基苯等,因其氧化电势高,不能向复合体供电子,因此,不能通过电子转移途径被氧化,而带有供电子基团(羟基、酯基)的有机物如苯酚可以被有效降解[29]。对于带有不同官能团的酚类化合物,可以通过循环伏安法检测其半波电位,通过构建反应速率与半波电位之间的QSAR 分析(图3(a)),从而确定其能否通过非自由基电子转移路径降解。
5.3 催化机理综合分析
对于一个机理未知的过硫酸盐催化体系,通过合理设计检测流程可以快速判定反应中生成的ROS 及其催化作用。首先,以DMPO 作为电子自旋捕获剂,通过EPR 测试分析体系中是否存在羟基自由基或硫酸根自由基,若EPR 测试检测到自由基的存在,则进一步通过甲醇、乙醇或叔丁醇淬灭验判断自由基在催化反应中的贡献率。需要注意的是,有机溶剂的添加量不宜过高,一般是过硫酸盐浓度的100~2 000 倍,因为过硫酸盐在甲醇等溶剂中的溶解度较低,加入过多会导致过硫酸盐的析出,从而影响反应机理的判断。若甲醇等淬灭剂对污染物降解反应无明显压制作用,这可能是只有少量的活性自由基生成,但是在催化中不起主要作用。除此之外,还可能是生成的活性自由基主要富集在碳材料的疏水表面,甲醇等亲水性淬灭剂难以清除。为了判别是否存在固体表面自由基,此时应当选择1~2 种难降解的缺电子有机污染物如苯甲酸或硝基苯作为探针分子进行降解测试,由于这些有机污染物的氧化电势较高,不能被非自由基途径所降解。若该催化体系不能被甲醇等淬灭剂压制,而能降解苯甲酸等缺电子有机物,说明固体表面自由基在催化中起到主要作用。此外,还可以通过荧光光谱法半定量分析反应中自由基的生成情况。
若以上机理分析手段发现反应体系中没有活性自由基生成,这说明反应体系可能存在非自由基途径,即体系可能产生1O2或发生电子转移反应。此时,应以TEMP 作为电子自旋捕获剂,通过EPR 观测是否有代表1O2的1 ∶1 ∶1 三重峰存在。需要说明的是,EPR 测试只能定性分析活性自由基和1O2是否产生,不能确定它们是否参与了反应,需要淬灭实验或溶剂交换实验来确定。若检测到1O2信号,则在反应体系中加入化学淬灭剂(FFA、TEMP 等)或使用重水为溶剂进行反应,通过对比污染物降解率的变化来确定1O2的贡献率。最后通过开路电位法、原位Raman 法和选择性氧化等方法来确定是否存在电子转移过程及其在催化中的相对贡献。
6 结论与展望
过硫酸盐高级氧化技术近年来得到了快速发展,碳纳米管材料作为一种新型、绿色、高效的碳基催化剂,已在水处理领域展现出优异的性能和应用前景。CNT 及其复合材料不仅能够使过硫酸盐的O—O 键断裂,产生活性自由基和单线态氧,还可以吸附过硫酸盐使表面氧化电势抬升,通过电子转移途径选择性降解CNT 表面共吸附的富电子有机污染物。尽管目前关于碳纳米管催化的应用已取得一定的成效,但现有研究仍存在一些亟待解决的问题:
(1)天然水体成分复杂,有多种有机物、重金属、天然有机质和阴离子等共存,可能会影响碳纳米管催化的界面行为和机理。当前的研究大多是在实验室环境下所做的针对单一污染物的降解,其浓度水平和溶液条件与实际水体环境有很大的差异。因此,未来要更加关注实际水体以及复合污染研究。
(2)基于碳纳米管活化过硫酸盐高级氧化技术虽然可以快速去除污染物,但是仍有部分中间产物矿化不完全,不仅会吸附在催化剂表面引起失活,而且中间产物的毒性可能比原始污染物的更强。因此,需要研究污染物的降解路径及中间产物毒性,并开发更多具有高矿化能力的限域催化材料和单原子催化剂,从而产生更多自由基和非自由基ROS 深度降解有机污染物。
(3)碳纳米管材料具有纳米尺寸,使得它们在实际环境中难以回收利用和管理控制,因而其应用受到限制,未来需要开发更多纳米器件化材料如三维气凝胶、纳米纤维和膜反应器等用于高级氧化技术中。
总之,碳纳米管作为高效的过硫酸盐催化剂,在水体深度净化领域中有广阔的发展前景和应用价值。
猜你喜欢
云南化工(2021年5期)2021-12-21
云南化工(2020年11期)2021-01-14
四川冶金(2019年5期)2019-12-23
经济技术协作信息(2018年30期)2018-11-22
浙江大学学报(工学版)(2016年9期)2016-06-05
中国资源综合利用(2016年1期)2016-02-03
合成化学(2015年4期)2016-01-17
分析测试学报(2015年8期)2016-01-13
原子与分子物理学报(2015年3期)2015-11-24
化工生产与技术(2014年6期)2014-02-27
