
基于DMC 法的α-氨基酸-N-环内酸酐的绿色合成
2023-11-08 17:58李振环彭日丽苏坤梅
天津工业大学学报 2023年5期
李振环,彭日丽,苏坤梅
(1.天津工业大学材料科学与工程学院,天津 300387;2.天津工业大学化学工程与技术学院,天津 300387)
氨基酸聚合物具有良好的降解性和生物相容性,是有极大发展潜能的生物医用高分子材料[1],作为药物递送载体[2]、生物分子沉淀剂[3]、抑菌材料[4]等在医药化学领域被广泛应用。α-氨基酸-N-环内酸酐(NCAs)是合成多肽的重要中间体,利用NCAs 可快速简便地合成固定序列的聚氨基酸[5],因此,NCAs 的合成研究被广泛关注[6-7]。
Boiteau 等[8]以N-氨基甲酰胺为原料发生亚硝化反应,失去氮气和水后得到L-缬氨酸-N-羧基环内酸酐(Val-NCA),产率高达95%。Semple 等[9]发现在N2气氛下,γ-苄基-L-谷氨酸加入三光气反应后,在乙酸乙酯与正己烷中重结晶得到的NCA 收率可达85%。Wang 等[10]在氨基酸-四氢呋喃悬浮液中加入双光气后,用硅藻土去除副产物氯化氢,可规模产出多种NCAs。Koga 等[11]以两性离子形式将3 种氨基酸转换为对应的咪唑盐后,在乙腈中与碳酸二苯酯结合生产氨基甲酸酯衍生物,最后加入甲酸环化成NCAs,产率在75%以上。但上述方法均有毒性大、对环境不友好的缺点[12]。碳酸二甲酯(DMC)作为一种无毒绿色试剂,具有化学活性高和廉价易得的优点[13],是一种良好的甲氧羰基化试剂[14],是代替光气合成NCAs 的更好选择。本课题组早前将DMC 作为环化剂,尝试了N-羧基丙氨酸酸酐(Ala-NCA)[15]与L-谷氨酸-γ-苄酯-N-羧酸酐(BLGNCA)[16]的合成,有产率较低或操作复杂的局限性。
基于以上问题,本文以乙酸锌为催化剂,采用“一锅法”合成L-苯丙氨酸-N-羧基环内酸酐(Phe-NCA)、Nε-苄氧羰基-L-赖氨酸环内酸酐(Cbz-Lys-NCA)和L-天冬氨酸-4-苄酯-羧基环内酸酐(H-Asp-Obzl-NCA),考察反应温度、反应物比例等因素对氨基酸转化率与NCAs 产率的影响,并对乙酸锌在反应过程中的催化作用进行分析,以期为氨基酸聚合物的绿色合成提供参考。
1 实验部分
1.1 原料、试剂与设备
主要原料与试剂:苯丙氨酸(Phe)、L-天冬氨酸-4-苄酯(H-Asp-Obzl)、Nε-苄氧羰基-L-赖氨酸(Cbz-Lys)、无水乙酸锌、无水碳酸钠,均为上海麦克林生化科技股份有限公司产品;N,N-二甲基甲酰胺(DMF)、DMC,均为天津科密欧化学试剂有限公司产品,使用前采用4A 分子筛进行除水处理。
设备:LC-210 型高效液相色谱仪,安捷伦科技(中国)有限公司产品;ELSD-2000 型蒸发光散射检测器,美国奥泰科技(中国)有限公司产品;AVANCE III HD 400 MHz 型液体核磁共振波谱仪、D8 ADVANCE型X-射线衍射分析系统,均为德国布鲁克公司产品;NEXSA 型X 射线光电子能谱仪,Nicolet Is 50 型傅里叶红外光谱仪,均为美国赛默飞科技有限公司产品;SCION 456-TQ 型气相色谱-质谱联用分析系统,德国SCION 仪器有限公司产品。
1.2 α-氨基酸-N-环内酸酐的制备
α-氨基酸-N-环内酸酐的合成路线如图1 所示。

图1 NCAs 的合成路线Fig.1 Synthetic route of NCAs
分别称取5 mmol 的氨基酸(Phe 0.825 g、H-Asp-Obzl 1.11 g、Cbz-Lys 1.40 g)加入三口烧瓶中,再加入50 mL 处理后的DMF 溶剂,在冷凝回流的条件下加热搅拌,使氨基酸在适宜温度下溶解。称取7.5 mmol(0.795 g)的Na2CO3和1 mmol(0.18 g)Zn(OAc)2加入三口烧瓶,再倒入一定体积的DMC,通N2去除三口烧瓶中的空气,再用生料带密封瓶塞处,继续加热2~10 h。反应结束后,趁热抽滤得到澄清溶液,再先后加入适量的正己烷与乙酸乙酯,密封置于-2 ℃下48 h,析出淡黄色固体,过滤,冷冻干燥后即得到产物NCAs。最后对反应滤液进行减压蒸馏,并通过水洗萃取,得到反应后的催化剂,并将反应前后结构及形貌变化进行对比。
在碳酸钠提供的碱性环境下,氨基酸中的氨根离子实现去质子过程,再与DMC 中的甲氧羰基进行亲核反应后完成羰基化。然后,在乙酸锌中Zn2+的作用下,发生分子内亲核取代反应,脱去一分子CH3OH[17],环化合成NCAs。两步反应均可在“一锅法”条件下进行,实现NCAs 合成[18]。
1.3 性能测试与结构表征
(1)氨基酸转换率与NCAs 产率测定:采用LC-210 型高效液相色谱仪连接ELSD-2000 型蒸发光散射检测仪组成高效液相色谱-蒸发光散射(HPLCELSD)检测系统,以标准曲线法定量分析氨基酸的转换率与NCAs 产率。液相测试条件为流动相100%HPLC-DMF,流量0.6 L/min;蒸发光散射测试条件为喷嘴温度100 ℃,N2流速3 L/min。
(2)气相色谱-质谱联用分析:采用SCION 456-TQ 型气相色谱-质谱联用分析系统。气相测试条件为:N2流速0.5 L/min,进样口温度250 ℃,温度以10 ℃/min 速率由50 ℃升至200 ℃保持5 min,再以同样的升温速率升至250 ℃,保持10 min。质谱测试条件为:离子化能量70 eV,离子源温度200 ℃。
(3)傅里叶红外变换表征:采用Nicolet Is 50 型傅里叶红外光谱仪,将样品于60 ℃烘干后,与KBr 以一定比例混合后研磨压片进行测试,测试波数为500 ~4 500 cm-1。
(4)液体核磁共振波谱表征:采用AVANCE III HD 400 MHz 型液体核磁共振波谱仪。将产物溶解于氘代二甲亚砜,对其进行1HNMR 和13CNMR 检测。
(5)X 射线光电子能谱表征:在室温条件下采用NEXSA 型X 射线光电子能谱仪对样品进行XPS 表征,测试步长为0.1 eV。
(6)X 射线衍射表征:采用D8 ADVANCE 型X-射线衍射分析系统。Cu-Kα 为辐射源,扫描角度为5°~80°,扫描速率为5°/min,工作电流为40 mA,电压为40 kV。
2 结果与讨论
2.1 α-氨基酸-N-环内酸酐的定性分析
2.1.1 气相色谱-质谱联用表征
图2 所示为NCAs 的GC-MS 谱图。其中,图2(a)为氨基酸与DMC 反应后上清液的GC 谱图;图2(b)—图2(d)为对应时间所得的质谱图。
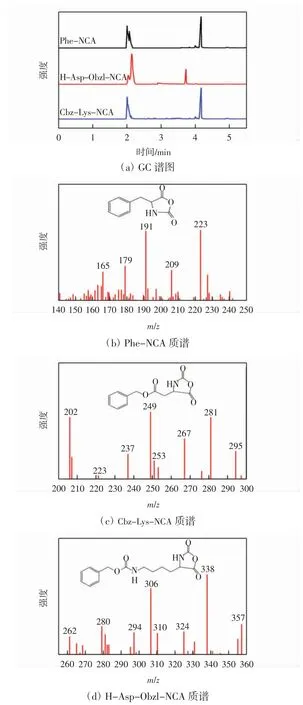
图2 NCAs 的GC-MS 谱图Fig.2 GC-MS spectrums of NCAs
由图2 可知,Phe-NCA、H-Asp-Obzl-NCA、Cbz-Lys-NCA 的气相峰分别出现在4.18、3.72、4.15 min,峰形与位移时间同标准样品基本一致,其对应相对分子质量分别为191、249 与306。甲氧羰基化后的中间体相对分子质量分别对应223、281 与338。通过相对分子质量换算可发现,3 份反应液中,除NCAs 产物外,均存在未反应的氨基酸、未环化的中间体以及一定量的副产物,其结构式如图3 所示。例如,图2(b)中相对分子质量为179 的信号为氨基与DMC 分子上的CH3结合成的副产物:α-氨基酸-N-甲基化(图3 中的A)。由图2(c)和图2(d)中可以看出,除去部分副产物外,中间体上羧基也出现可能被二次甲基化的现象,出现甲氧羰基氨基甲酸酯的副产物(图3 中的B)。由于NCAs稳定性较差,还可能脱去一分子CO2,失去相对分子质量为44 的分子碎片。

图3 副产物的结构式Fig.3 Structural formula for by-products
2.1.2 傅里叶红外变换表征
图4 为NCAs 的FT-IR 谱图。3 种氨基酸的氨基脱去质子,与DMC 中的酰基结合成酰胺键,在催化剂的作用下转变为NCAs。
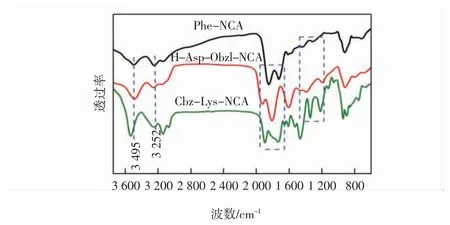
图4 NCAs 的红外表征Fig.4 FT-IR spectra of NCAs
由图4 Phe-NCA 的红外谱图中可以看出,在3 252 和3 495 cm-1处出现的是环内酰胺的特征吸收峰,在1 729 和1 855 cm-1附近,分别为环状酸酐中2个C—O 的伸缩振动峰,推断产物为环状酸酐。由Cbz-Lys-NCA 和H-Asp-Obzl-NCA 的图谱可更清晰地观察到1 300~1 200 cm-1间环状酸酐中C—O 键的伸缩振动峰,而1 340~1 300 cm-1处表现为酰胺的Ⅲ谱带,1 560~1 640 cm-1为N—H 的弯曲振动。由此证明DMC 法成功地制备出了3 种NCAs。
2.1.3 液体核磁共振波谱表征
图5 为NCAs 的核磁谱图。

图5 NCAs 的核磁氢谱图和核磁碳谱图Fig.51H NMR and 13C NMR spectra of NCAs
由图5 中的核磁氢谱图可知:化学位移δ=2.49处为溶剂氘代二甲亚砜所属信号。氨基酸酸酐环上的氨基质子的信号出现在化学位移δ=8.0 处。由于HAsp-Obzl-NCA 中COOC2H5的吸电子共轭作用,导致其略微向低场移动,并在δ=8.19 处出峰。Cbz-Lys-NCA 中酸酐环中和碳链上的亚甲基质子信号也在一定程度向低场移动,分别在5.10、3.61、3.01、2.91 和2.74处出峰,这是由—NH2强吸电子诱导所引起的。NCAs的其余官能团的化学位移均与其标准核磁氢谱相吻合。对比图5 中核磁碳谱图可以发现,酸酐环中O—C—O—C—O 的化学位移出现在谱图中的167.0 和157.5 处,这进一步证明了NCAs 的成功合成。
2.2 反应条件的考察
2.2.1 去质子碱及乙酸锌用量对反应的影响
去质子碱与催化剂的用量比对氨基酸转化率(C)以及NCAs 产率(Y)的影响如表1 所示。
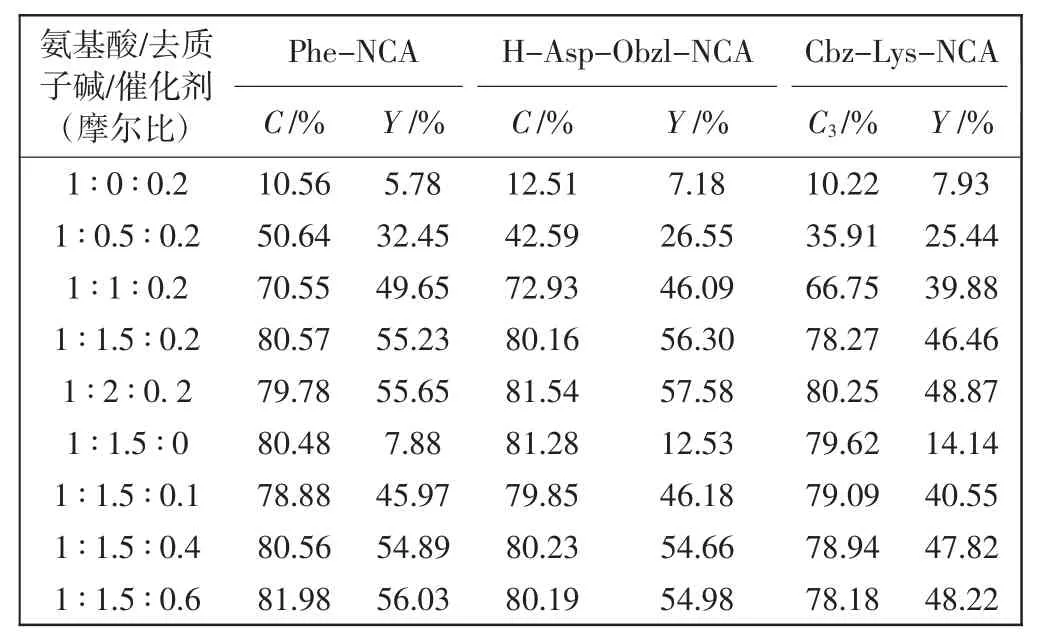
表1 去质子碱及乙酸锌用量对反应的影响Tab.1 Effects of dosage of deprotic alkali and zinc acetate on reaction
由表1 可知,当反应体系中不加入Na2CO3时,氨基酸转化率只有10%左右。这是由于常温下氨基酸一般以氨基夺取羧基质子、氨基带正电、羧基带负电的两性离子形式存在,不易发生反应。通过去质子化才能使氨基酸与DMC 反应,实现氨基酸的转换。不同于强碱试剂,Na2CO3作为弱碱,能在不改变氨基酸活性的情况下,中和氨基上的质子,使氨基酸进入反应活化状态。同时利用Na+与羧基相连,一定程度可以阻止DMC 上的活性基团与其相连。表1 中数据证明,当氨基酸与去质子碱的摩尔比为1 ∶1.5 时,3 种氨基酸的转化率均在80%左右,继续增加Na2CO3的比例,转化率变化不大。由表1 可以看出,当反应体系中不添加乙酸锌催化剂时,NCAs 的选择性降低,使得产率下降,Phe-NCA 在不添加乙酸锌的情况下,产率仅为7.88%。当氨基酸与催化剂的摩尔比为1 ∶0.2 时,NCAs选择性最高,Phe-NCA、H-Asp-Obzl-NCA 和Cbz-Lys-NCA 的产率分别为55.22%、56.30%、46.46%,继续增加乙酸锌用量,产率没有明显增加,意义不大。综上可知,反应中氨基酸∶去质子碱∶催化剂的摩尔比为1 ∶1.5 ∶0.2 最为合适。
2.2.2 反应温度对催化反应的影响
反应过程中氨基酸的溶解度、中间体的成环及裂解均与反应温度有着密切的联系。以20 ℃为梯度,研究温度对NCAs 的产率与选择性的影响,结果如图6 所示。
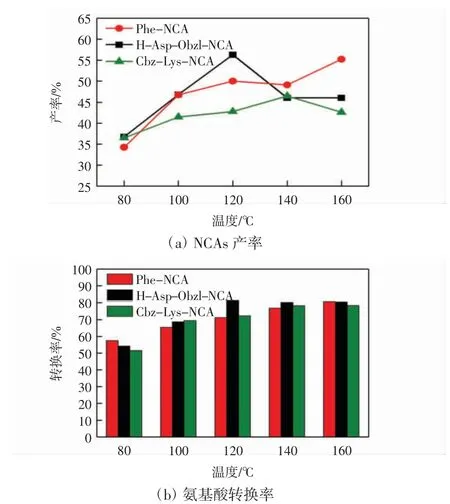
图6 温度对NCAs 产率与氨基酸转换率的影响Fig.6 Effect of temperature on conversion rate and yield of NCAs
由图6 可知,由于氨基酸结构差异,不同NCAs 的最适宜反应温度有所差异。Phe-NCA 在160 ℃时转换率最高,为80.58%,能达到最高产率55.22%。H-Asp-Obzl-NCA 在120 ℃时转化率可以达到81.36%,产率为56.30%。Cbz-Lys-NCA 最佳反应温度为140 ℃,转化率为78.27%,能达到最高产率46.46%。随着温度的提升,氨基酸在DMF 中的溶解度增大,参与反应的原料增加,产率和选择性逐渐变大。3 种氨基酸中Phe 的溶解温度相对较高,因此,最佳反应温度也相应增高。超过反应的最佳温度后,NCAs 开始出现开环聚合,同时DMC 中也存在甲基化活性位点,导致氨基酸容易出现甲基化反应,因此,随着温度提高,H-Asp-Obzl-NCA 和Cbz-Lys-NCA 的产率出现先增后减的趋势。
2.2.3 反应物间比例对催化反应的影响
反应物间摩尔比对NCAs 产率与氨基酸转换率的影响如图7 所示。
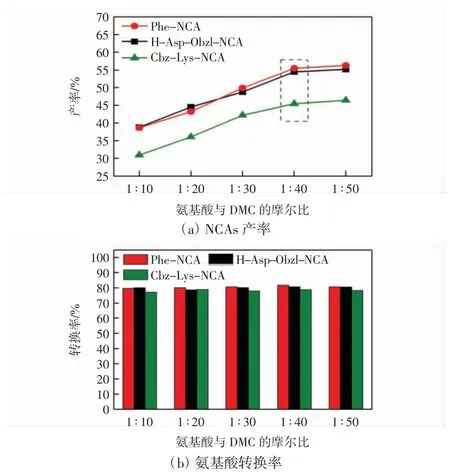
图7 反应物间摩尔比对NCAs 产率与氨基酸转换率的影响Fig.7 Effect of molar ratio between reactants on conversion rate and yield of NCAs
氨基酸与DMC 间的摩尔比也是影响NCAs 产率的关键因素。由图7 可知,由于DMC 的沸点较低,而反应最佳温度均在120 ℃以上,即便在冷凝回流的条件下DMC 也无法全部活化参与反应,因此,增加体系中DMC 的含量时,更多的C—O 键的氧与羰基碳形成共轭的平面结构[19],与乙酸锌上的Zn2+结合后,对氨基酸进行甲氧羰基化反应,导致氨基酸转化率升高;同时由于Zn2+的强吸电子能力,使得氨基酸上的—COO—与羰基碳结合,实现环化,故NCAs 的产率明显提升。当氨基酸与DMC 的摩尔比达到1 ∶40 时,NCAs 产率逐渐稳定。
2.3 催化剂作用分析
为了证实催化剂乙酸锌在反应过程中发挥的作用,对反应前后的乙酸锌进行表征。
2.3.1 SEM 表征
乙酸锌作为均相催化剂,需减压蒸馏并萃取后加入沉淀剂分离。图8 为催化剂反应前后的SEM 对比图。

图8 Zn(OAc)2 反应前与反应后的扫描电镜图Fig.8 SEM images of Zn(OAc)2 before and after reaction
由图8 可见,未参与反应的乙酸锌为不规则片层状结构;反应结束后,乙酸锌的片层结构消失,回收的催化剂尺寸变大且表面变得光滑。这是由于分离催化剂过程中乙酸锌团聚以及溶剂残留所导致的。
2.3.2 XPS 表征
为了进一步证实催化剂在反应过程中发挥的作用,对反应前后Zn(OAc)2中的Zn 和O 元素进行了光电子能谱测试,结果如图9 所示。

图9 Zn(OAc)2 反应前和反应后的X 射线光电子能谱分析Fig.9 X-ray photoelectron spectroscopy of Zn(OAc)2 before and after reaction
由图9 中Zn2p 反应前后的分峰图可以看出,Zn2p3/2的结合能由1 021.9 eV 增加至1 025.7 eV。这是由于乙酸锌中原本亲电的Zn2+进攻羰基上富电子的氧[20],与DMC 中的羰基氧进行配位络合为Zn—O[21]键后,其周边的电子密度有所下降。反应过程中,去质子后的氨基作为亲核试剂[22],进攻DMC 上的羰基碳原子发生甲氧基羰基化反应,导致乙酸锌和DMC 配合物中的配位键Zn—O 断裂。然后乙酸锌中的Zn2+再与氨基甲酸酯中的C—O 配位,使氨基甲酸酯中的羰基亲电性增强,进而使得COONa 或COOH 中的O 容易进攻氨基甲酸酯中的羰基,实现精准环化。O1s 分峰图在528~535 eV有ZnO 的特征峰,证明反应过程中乙酸锌部分水解。
2.3.3 FT-IR 表征
乙酸锌反应前后的FT-IR 谱图如图10 所示。

图10 催化剂反应前后的FT-IR 谱图Fig.10 FT-IR spectra of catalyst before and after reaction
由图9 可知,参与反应前的乙酸锌的FT-IR 谱图在1 465 和1 565 cm-1处有峰,其分别为乙酸根离子的对称伸缩峰与不对称伸缩峰,而反应后的催化剂则在1 437 和1 600 cm-1处出峰。对比反应前后,会发现两峰的间距明显变大。这是由于Zn2+与DMC 结合后使得乙酸锌由双齿配合物转变为单齿配合物所导致的[19]。同时,反应后的催化剂在524 cm-1处出现了Zn—O 的伸缩振动峰,也说明催化剂部分失活。在614 和706 cm-1处的峰则为催化剂表面残留的中间体中酰胺的Ⅴ和Ⅳ谱带,进一步说明乙酸锌参与了反应,催化氨基酸实现甲氧羰基化过程。
2.3.4 XRD 表征
将反应前后的催化剂进行XRD 表征,结果如图11 所示。
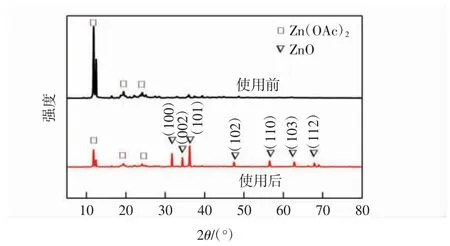
图11 反应前和反应后Zn(OAc)2 的X 射线衍射图Fig.11 XRD pattern of Zn(OAc)2 before and after reaction
由图11 可以看出,反应后的乙酸锌在11.79°、19.49°、25.73°处的衍射峰减弱,但在30°~70°范围内出现了新的衍射峰,该衍射峰归属于ZnO 的(100)、(002)、(101)、(102)、(110)、(103)、(112)等晶面衍射(标准卡JCPDS No.01-0089),证明乙酸锌在反应过程中或回收过程中发生了水解。乙酸锌水解是造成NCAs 产率下降的主要因素,开发合成一种非均相且有效大幅度提升酸酐产率的催化剂将是后续进一步探讨的课题。
综上所述,推测催化剂乙酸锌在反应过程中的作用机理为:Zn2+与DMC 中的羰基氧进行配位络合为Zn—O 键,使得电子云偏离羰基碳,更易与氨基反应得到氨基甲酸酯。氨基甲酸酯中的羰基更容易与Zn2+配位,从而增强了氨基甲酸酯中羰基的亲核能力,并促进邻近COO-与羰基碳发生亲核取代,环化生成NCAs。
3 结 论
以氨基酸为原料、乙酸锌为催化剂、DMC 为环化剂,通过“一锅法”高效环保地合成了Phe-NCA、HAsp-Obzl-NCA 和Cbz-Lys-NCA 3 种新型NCAs。研究结果表明:
(1)在乙酸锌的催化下,采用“一锅法”成功合成了3 种新型α-氨基酸-N-环内酸酐(Phe-NCA、HAsp-Obzl-NCA、Cbz-Lys-NCA)。
(2)在体系中氨基酸∶去质子碱∶催化剂的摩尔比为1 ∶1.5 ∶0.2、氨基酸与DMC 摩尔比为1 ∶40 的条件下,反应进行8 h,Phe-NCA 在160 ℃下产率可达到55.22%。H-Asp-Obzl-NCA 在120 ℃下达到最佳产率56.30%,Cbz-Lys-NCA 在140 ℃的产率为46.46%。
(3)乙酸锌与DMC 的配位络合现象和电子密度偏差,促进DMC 上的甲氧羰基的转移和中间产物发生亲核取代,从而完成环化得到NCAs。
猜你喜欢
陶瓷学报(2021年5期)2021-11-22
国际放射医学核医学杂志(2020年4期)2020-07-27
高中数理化(2016年19期)2016-11-14
中国塑料(2016年2期)2016-06-15
航天制造技术(2016年6期)2016-05-09
中国塑料(2016年5期)2016-04-16
云南中医学院学报(2015年1期)2015-07-31
应用化工(2014年1期)2014-08-16
太空探索(2014年6期)2014-07-10
生物加工过程(2013年1期)2013-03-11
