
基于RAA-CRISPR/Cas12a快速检测尼帕病毒方法的建立
2023-11-08 06:56王英丽张永强李金明包静月王志亮
中国动物检疫 2023年10期
徐 蛟,王英丽,王 莹,常 星,张永强,李金明,包静月,王志亮
(中国动物卫生与流行病学中心,山东青岛 266032)
尼帕病毒性脑炎是一种由尼帕病毒(Nipah virus,NiV)引起的人兽共患病。NiV主要感染马、羊、犬、猪等动物,亦可感染人类,易感动物病死率高达80%。20世纪90年代,尼帕病毒性脑炎疫情首次在马来西亚暴发,数百人被感染,死亡率接近50%,另有数百万头猪在此次疫情中被扑杀,给当地养殖业造成了极大损失[1-2]。随后该病在东南亚呈区域性流行态势[3-5]。动物和人感染后主要表现为发热、头痛、咳嗽、呼吸困难以及腹泻,严重者会出现病毒性脑炎以及癫痫症状[6]。虽然目前该病尚未传入我国,然而鉴于其危害性,我国将其列为一类动物疫病。NiV对养殖业安全以及公共卫生安全危害极大,当下世界范围内尚无有效疫苗和治疗药物。尽管国内尚未报道尼帕病毒性脑炎病例,然而马来西亚、孟加拉国等均出现了相关病例,且作为该病毒天然宿主的果蝠在我国南方省份也有自然分布,这些因素均提示该病的传入风险较大。
NiV是一种单股负链RNA病毒,属于副黏病毒科亨尼帕病毒属,全长约18.2 kb,直径126~150 nm,电镜下观察其主要呈球形和细长形。病毒RNA编码6种结构蛋白(N、P、M、F、G、L),其中糖蛋白(G)和融合蛋白(F)负责病毒与受体的结合。结构蛋白中磷蛋白(P)参与病毒RNA初始转录,大蛋白(L)和核蛋白(N)参与病毒复制,基质蛋白(M)介导病毒组装和病毒颗粒释放。
鉴于尼帕病毒性脑炎的高死亡率,对其快速诊断在临床实践中显得尤为重要。目前已经建立的检测方法虽然灵敏度较高,但是操作步骤复杂,十分依赖专业操作人员以及精密仪器设备,无法满足现场对NiV的快速检测,所以建立一种快速、准确、易操作的检测方法对于早期发现NiV十分必要。本试验建立了基于RAA-CRISPR/Cas12a的NiV检测方法,其不需要精密的仪器设备,在常温条件下即可实现对NiV的快速检测,为NiV的早期发现并及时调整应对措施提供了技术支撑。
1 材料与方法
1.1 病毒阳性质粒
鉴于国内尚无尼帕病毒性脑炎病例,也无法获取病毒核酸,因此本方法中涉及NiV的检测均以构建的病毒质粒为基础。根据crRNA序列选取原则(见1.3),选取NiV(NC002728.1)N和F基因相应区域合成片段,并连接至pUC57载体,作为后续试验的NiVF/N基因阳性质粒。随机选取NCBI中收录的3个NiV序列(MK575067.1、MK673578.1、FJ513078.1),根据上述原则选取N基因序列和F基因中crRNA结合区域,并合成相应片段连接至pUC57载体,为后续特异性试验备用。

图1 NiV-RAA引物的扩增结果
1.2 主要试剂和仪器
CRISPR/Cas12a,购自上海吐露港生物科技有限公司;RAA试剂盒,购自山东赛恩斯科技有限公司;DNA提取酚试剂盒,购自北京索莱宝科技有限公司;pUC57-NiV-N和pUC57-NiV-F阳性质粒,由上海生工生物公司构建。实时荧光定量PCR仪,购自Bio-Rad公司;NanoDrop-2000超微量紫外分光光度仪,购自美国Thermo Fisher公司。
1.3 crRNA、RAA引物以及阳性质粒设计合成
从NCBI下载NiV序列进行比对,寻找病毒基因序列保守区域,并在保守区域内寻找TTTN(N = A,T,G)序列,然后向后选取21 nt作为备选序列,Blast分析该序列特异性。经过筛选,分别在病毒N和F基因上发现符合要求的TTTN序列,在该序列前加上固定的发卡结构序列即形成最终crRNA序列。在选定的TTTN序列前后位置分别选取保守序列,根据RAA引物设计原则,利用Premier 5软件设计NiV RAA引物,根据上述原则,分别将N和F基因含有crRNA结合区域以及其前后RAA上下游引物结合的保守区域,作为合成质粒的片段构建阳性质粒。设计一段12 nt随机序列的ssDNA,分别在序列两端加上荧光基团与淬灭基团作为CRISPR/Cas12a反应体系中的报告DNA,序列信息详见表1,上述所有序列均由上海生工生物公司合成。分别将含有上述TTTN序列的N和F基因保守序列连接至pUC57载体,将经测序鉴定正确的质粒作为质粒标准品,分别命名为pUC57-NiV-N和pUC57-NiV-F。该质粒由上海生工生物公司构建和验证。
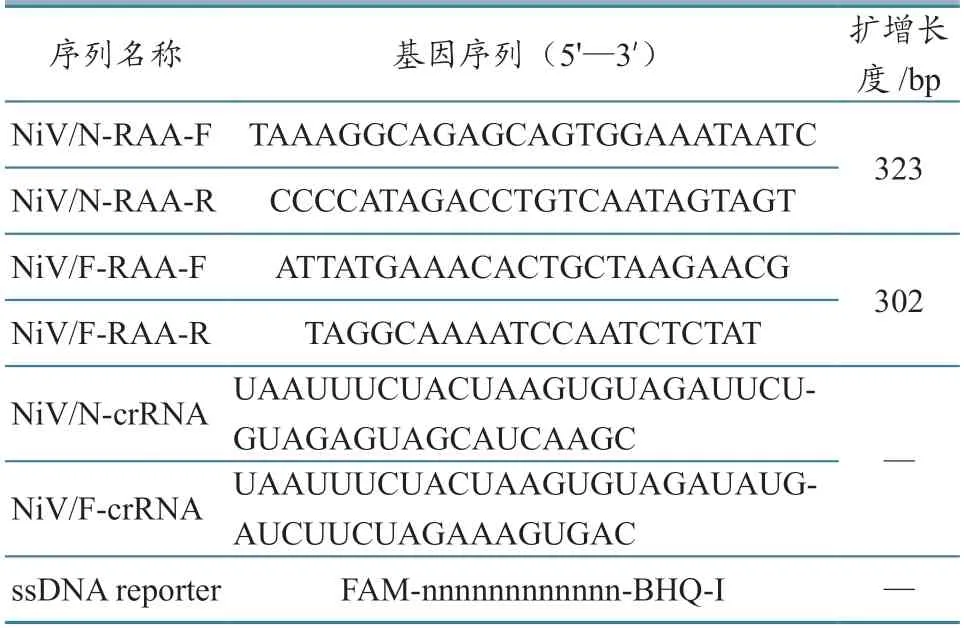
表1 RAA引物、crRNA以及报告DNA序列
1.4 RAA扩增及CRISPR/Cas12a酶检测
分别使用相同浓度的NiV阳性质粒模版(107拷贝/μL),通过RAA扩增以及后续的CRISPR/Cas12a系统,对设计的RAA引物的有效性以及crRNA引导Cas12a剪切ssDNA的能力进行验证。RAA反应体系为50.0 μL,其中A buffer 29.4 μL,上下游引物(10 μmol/L)各2.0 μL,核酸模板10.0 μL,DEPC水4.1 μL,最后向反应管中加入2.5 μL B buffer。充分混合反应管,37 ℃反应45 min,反应结束后向反应管中加入50 μL酚/氯仿(v:v= 1:1),抽提10 min;12 000 r/min离心5 min,取5 μL上清液进行核酸凝胶电泳检测,将扩增产物送至上海生工生物公司测序,验证所设计的RAA引物能否成功扩增出目的片段。CRISPR/Cas12a的反应体系为20 μL,其中吸取RAA的反应产物5 μL作为模板,HOLMES Buffer 2 μL,Cas12a(10 μmol/L)1 μL,cRNA(10 μmol/L)1 μL,ssDNA reporter(5 μmol/L)1 μL,最后加入ddH2O补足至20 μL,阴性对照将模板替换为DEPC水,阳性对照使用试剂盒内提供的阳性crRNA以及阳性模板。使用荧光定量PCR仪在37 ℃条件下反应20 min,检测通道为FAM,每分钟对反应管内的荧光强度进行读取,对两条crRNA的活性进行验证。
1.5 敏感性检验
使用Nanodrop 2 000分光光度计,对构建好的pUC57-NiV-N和pUC57-NiV-F阳性质粒进行质量浓度测定。换算公式:拷贝数(拷贝/μL)=[6.02×1023×质粒质量浓度(ng/μL)×10-9]/(片段长度×660)。经计算,得出拷贝数并进行梯度稀释至100~108拷贝/μL。以稀释后的阳性质粒为模板,分别利用两对构建的RAA引物和crRNA进行RAA扩增和CRISPR/CAS12a检测,使用荧光定量PCR仪在37 ℃条件下反应20 min,检测通道为FAM,每分钟对反应管内的荧光强度进行读取,对检测方法的敏感性进行验证。
1.6 特异性检验
以口蹄疫病毒(FMDV)、猪细小病毒(PPV)、猪圆环病毒2型(PCV-2)等临床上常见的猪病毒核酸提取物以及NiV质粒(107拷贝/μL)为模板,对所建立检测方法的特异性进行验证。
1.7 重复性检验
选取高(107拷贝/μL)、中(105拷贝/μL)、低(103拷贝/μL)3个浓度水平的NiV阳性质粒,进行3次重复性检测,对建立的检测方法的可重复性进行验证。
2 结果
2.1 RAA扩增以及CRISPR/Cas12a检测
两对RAA引物的扩增结果(图1)显示:37 ℃水浴条件下扩增45 min后,含有两条crRNA互补配对的核酸检测靶标条带均被成功扩增,大小分别为323和302 bp。经过测序验证,扩增产物序列正确,表明设计的两对RAA引物均能够成功扩增目的片段。RAA-CRISPR/Cas12a反应体系的切割活性验证结果(图2~3)显示,在蓝光照射下,NiV/N-crRNA以及NiV/F-crRNA反应管在15 min后均出现了肉眼可见的荧光,证明Cas12a蛋白的反式切割活性被激活,并成功检测到构建的阳性质粒。荧光强度检测结果(图3)表明,两条crRNA均顺利结合到靶序列,激活ssDNA荧光信号,并在15 min内实现了对NiV阳性质粒的快速检测。
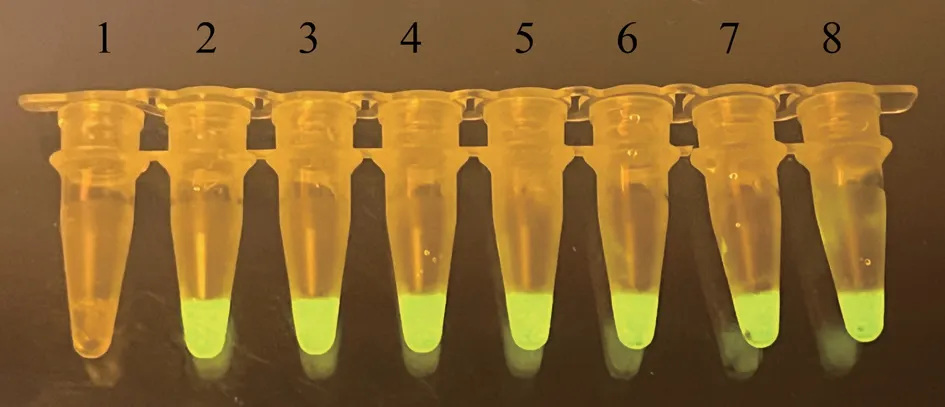
图2 蓝光下两条crRNA活性检测结果
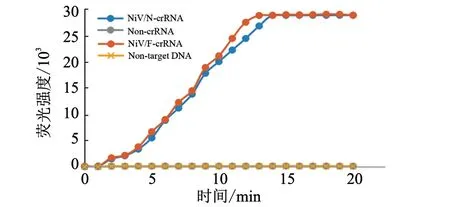
图3 两条crRNA活性检测结果
2.2 敏感性验证
将配制的不同浓度阳性质粒作为模版,进行敏感性试验。结果(图4)显示,CRISPR/Cas12a反应体系能够检测到102拷贝/μL的阳性质粒,且荧光信号强度在15 min内均达到高峰,而100~101拷贝/μL的阳性质粒无荧光信号。结果表明,构建的方法的检测下限为102拷贝/μL,灵敏性较高。
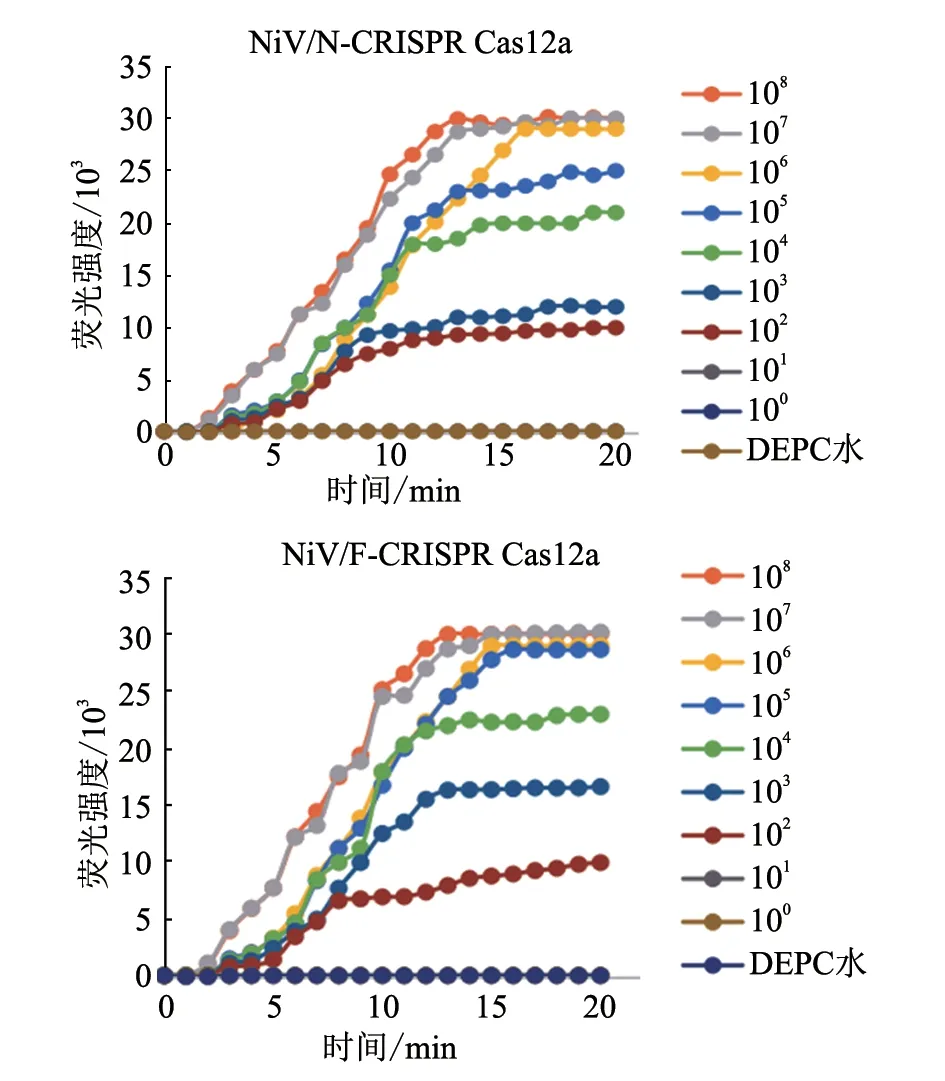
图4 敏感性验证结果
2.3 特异性验证
特异性检测结果(图5)显示,经RAA扩增后,CRISPR/Cas12a体系能够在15 min内完成对所有阳性质粒检测,并有检测信号出现,两条crRNA均能高效检出NiV阳性质粒,而对FMDV,PPV,PCV-2等猪属动物其他病毒均无交叉反应。结果表明,建立的方法特异性良好。
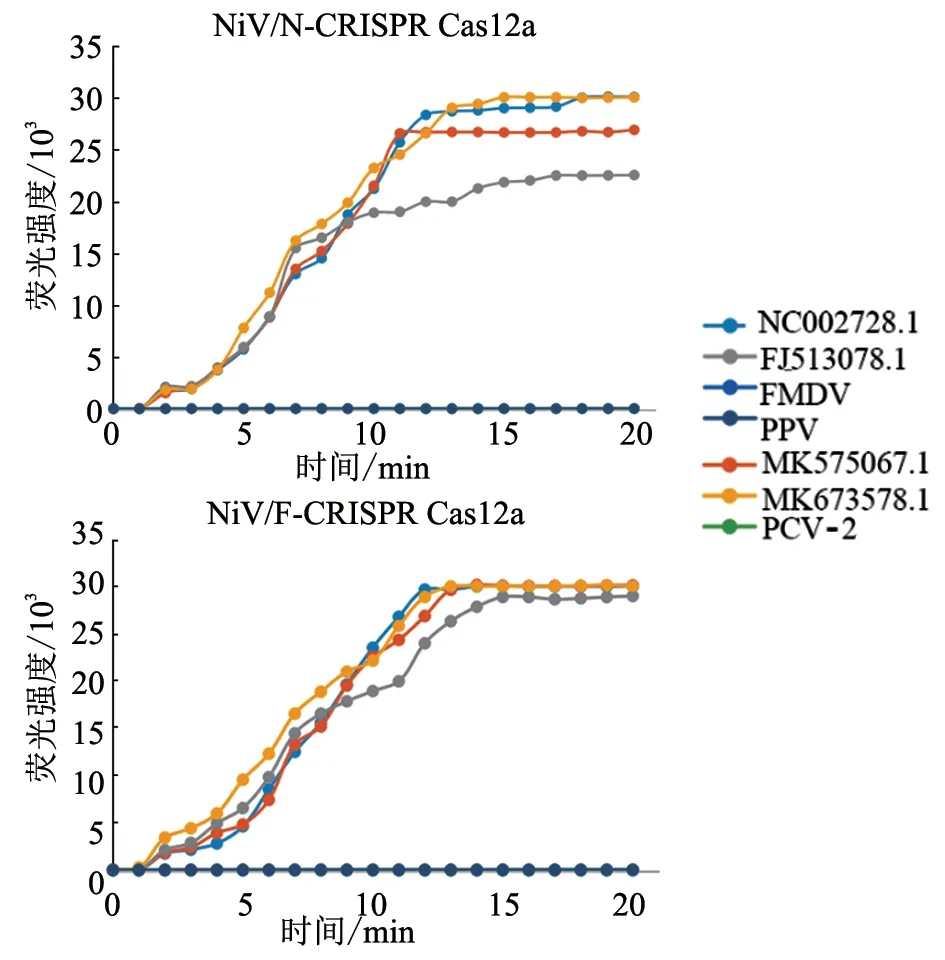
图5 特异性验证结果
2.4 重复性验证
高中低3个浓度水平的NiV阳性质粒重复性验证结果(图6)显示,对于不同浓度水平的阳性质粒,本研究所建立的方法均能够重复检出,且结果一致性良好。结果表明,该方法具有良好的重复性,稳定性较高。
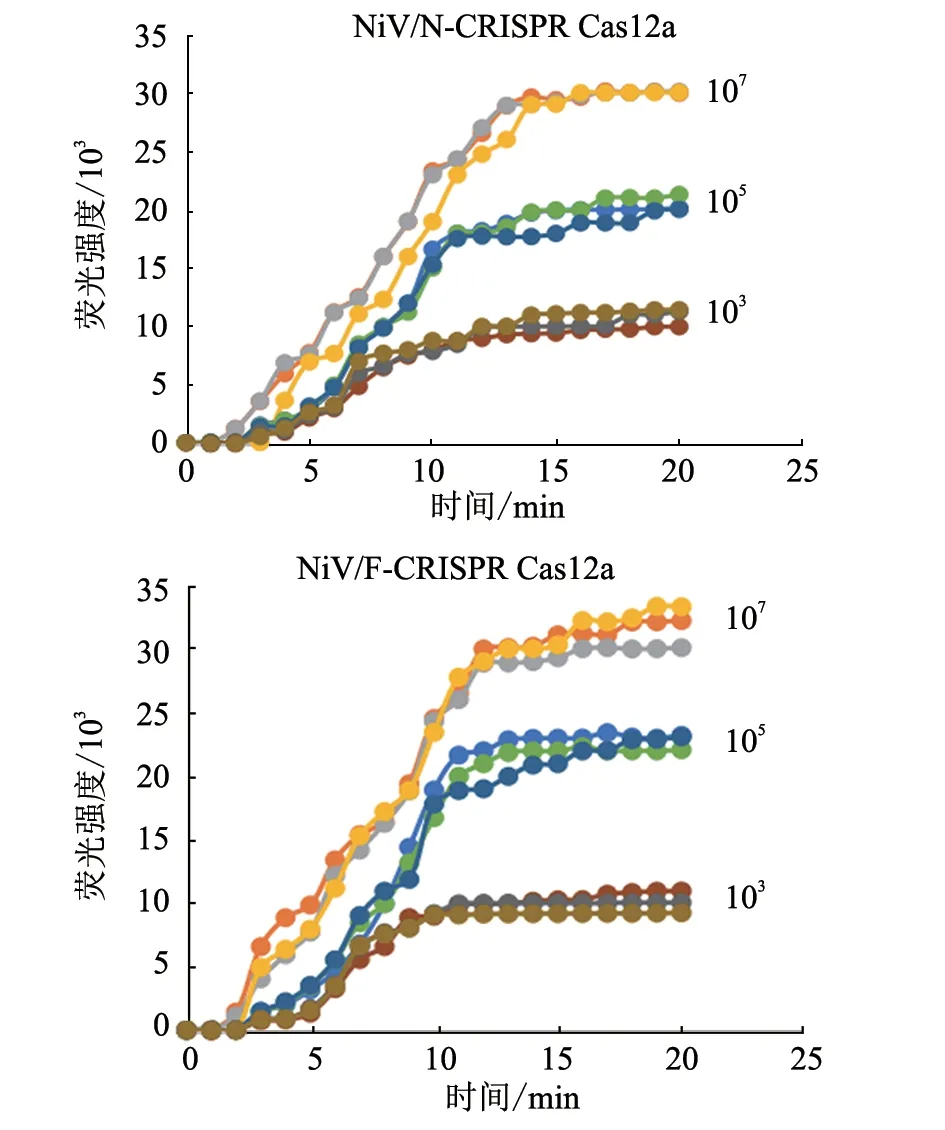
图6 重复性验证结果
3 讨论
本试验通过RAA-CRISPR/Cas12a技术成功建立了快速检测NiV的方法。鉴于引物设计时在NCBI上比对了上百条NiV序列,并筛选出保守区域中的靶序列,该方法对现有NiV检测适用性非常高,其在37 ℃条件下反应1 h即可完成对NiV阳性质粒的检测,且结果肉眼可见,这与传统RTPCR以及ELISA方法相比,节约了检测时间,简化了仪器设备,甚至在极端条件下,恒温水浴锅即可完成病毒检测全过程,完全能够用于田间诊断。该方法与其他常见猪病毒没有交叉反应,且具有较高的检测敏感性,稳定性较好,这对于基层工作者早期发现疫病进而采取措施阻断传播具有重要意义。
重组酶介导扩增(RAA)因其快速、简便、等温而成为现场诊断的首选扩增技术,但可能存在非特异性扩增的假阳性问题。规律成簇的间隔短回文重复序列及其相关蛋白(CRISPR/Cas)是原核生物中的一种适应性抗病毒免疫系统,存在于大多数细菌中,具有良好的特异性和可靠性[7]。Cas12a又称Cpf1,通过crRNA与特定的靶序列结合,进而激活CRISPR/Cas12a的反式切割活性,ssRNA reporter被切割后,其5'端的荧光基团和3'端的荧光淬灭基团分离进而发出荧光信号[8]。RAA与CRISPR的结合可以兼具灵敏度和特异性,在开发诊断技术方面具有广阔前景。
CRISPR/Cas12a已经被广泛应用于动物疫病诊断中。Jiang等[9]建立了基于ORF068基因诊断牛结节性皮肤病病毒(LSDV)的RPA-CRISPR/Cas12a诊断技术,相比较传统PCR和荧光RTPCR,新检测技术仅用时15 min即可完成检测,大大降低了检测周期,同时结果肉眼可见。Wang等[10]利用CRISPR/Cas12a成功构建了快速检测炭疽杆菌的检测系统,具有单拷贝级的灵敏度。Qian等[11]利用RAA-CRISPR/Cas12a成功建立起快速检测猪流行性腹泻病毒(PEDV)的方法,检测灵敏度达到102拷贝/μL,并在此基础上成功设计了使用更加便捷的试纸条,对于结果读取也更加便捷。
尽管与传统检测手段相比,CRISPR/Cas12a方法具备检测用时短、检测设备简单和对环境要求不高的优点,然而作为一种新兴技术,该方法存在的脱靶风险仍然不容忽视。crRNA需要通过PAM序列正确识别靶序列,这在增加了特异性的同时,也降低了靶区的选择和相应crRNA设计的灵活性,也导致了能够被用来当作靶序列的片段与传统方式相比大大减少[12]。此外,RNA酶的广泛存在导致crRNA非常脆弱,一旦操作不当极易造成RNA降解进而影响结果判定。随着研究的进一步深入,相信该技术在动物疫病诊断方面会有更加广阔的前景。
猜你喜欢
中国生殖健康(2018年1期)2018-11-06
食品科学(2018年10期)2018-05-23
中国医疗保险(2017年5期)2017-05-17
三峡大学学报(自然科学版)(2016年6期)2016-04-16
中国康复理论与实践(2015年10期)2015-12-24
西南医科大学学报(2015年1期)2015-08-22
现代电生理学杂志(2015年1期)2015-07-18
计算机与网络(2015年12期)2015-06-21
中国当代医药(2015年9期)2015-03-01
西南军医(2015年6期)2015-01-23
