
小儿特发性扩张型心肌病核心基因鉴定和免疫浸润分析
2023-11-07 11:33尤红俊赵倩倩苟棋玲董梦雅
中西医结合心脑血管病杂志 2023年20期
尤红俊,赵倩倩,苟棋玲,董梦雅
特发性扩张型心肌病(pediatric idiopathic dilated cardiomyopathy,PIDC)是儿童常见的心肌病,是造成儿童心力衰竭的主要原因之一,同时导致恶性心律失常、血栓栓塞和猝死在内的一系列不良结局。目前小儿扩张型心肌病(dilated cardiomyopathy,DCM)的早期诊治水平不断提高,但病因不明、缺乏精准治疗,此类患儿预后不容乐观[1-2]。本研究基于基因表达综合数据库(gene expression omnibus,GEO)中PIDC的二代测序数据,寻找疾病的差异表达基因(differentially expressed genes,DEGs),通过基因本体(Gene Ontology,GO)功能富集分析和京都基因与基因组百科全书(Kyoto Encyclopedia of Genes and Genomes,KEGG)通路富集分析探讨发病机制,并构建蛋白-蛋白互作(protein-protein interaction,PPI)网络,筛选其中的核心基因,整合其与心肌病及心力衰竭风险的关系,辅以免疫细胞浸润分析,寻找疾病发生发展过程中的关键基因,为疾病的精准靶向治疗提供依据。
1 资料与方法
1.1 数据下载及预处理
在GEO[3-4]中搜索“pediatric idiopathic dilated cardiomyopathy”,选取数据集GSE99321,其由美国科罗拉多大学药理学系Tatman P等科研人员免费提供,采用GPL16791 Illumina HiSeq 2500 (Homo sapiens)为平台,收录14例小儿左心室组织二代测序数据,其中7例小儿PIDC作为试验组,另7例无心力衰竭小儿作为对照组。数据进行归一化预处理。
1.2 差异表达分析
采用统计软件R语言(版本4.1.1)“DESeq2”[5]包进行差异基因表达分析。以|log2(fold change)|≥1,P<0.05为DEGs的筛选条件。运用R语言ggplot2和pheatmap包绘制火山图和热图,进行DEGs可视化分析。
1.3 GO功能富集和KEGG通路富集分析
使用R语言clusterProfiler和org.Hs.eg.db(Homo sapiens)包对DEGs进行GO功能富集分析和KEGG通路富集分析[6-7]。GO富集分析分别在生物过程(biological process,BP)、细胞组分(cellular component,CC)和分子功能(molecular function,MF)三方面对基因进行功能注释[8]。KEGG分析展示基因参与的信号通路。利用R语言ggplot2包将结果可视化。以P<0.05为差异有统计学意义。
1.4 PPI网络分析
将DEGs导入STRING(Search Tool for the Retrieval of Interacting Genes,http://string-db.org/cgi/input.pl),进行PPI网络分析[9]。设定可信度confidence为0.4,输出TSV格式文件,用于Cytoscape分析[10]。
1.5 核心基因筛选
使用Cytoscape 3.7.3 软件插件CytoHubba筛选出PPI网络中的核心基因。CytoHubba利用12种评分策略剖析PPI网络,包括最大相关准则(maximum correlation criterion,MCC)、度值(Degree值)等,该12种拓扑算法无优劣之分[11]。MCC代表基因/蛋白间最大相关性标准,以MCC值排序,居前5位的基因为PPI网络的核心基因。
1.6 利用CTD数据库评估核心基因与心肌病和心力衰竭的关联性
利用比较毒理基因组学数据库(comparative toxicogenomics database,CTD)(http://ctdbase.org/)分析核心基因与心肌病及心力衰竭风险的关系。CTD整合了化合物-基因/蛋白质相互作用、化合物-疾病和基因-疾病关系的信息,探索与疾病机制相关的假设[12]。
1.7 免疫细胞浸润分析
单样本基因集富集分析(single sample gene set enrichment analysis,ssGSEA)[13]根据表达谱计算每个基因的rank值,分析得到每个样本的免疫细胞的相对含量,有助于了解疾病状态下组织中所有免疫细胞的浸润情况,探讨其与疾病转归的关系。采用ssGSEA算法分析PIDC中28种免疫细胞在免疫浸润微环境中的比例。运用R语言pheatmap包和vioplot包绘制热图和小提琴图进行可视化免疫细胞浸润分析。以P<0.05为差异有统计学意义。
2 结 果
2.1 基因差异表达分析
对原始矩阵数据进行归一化处理(见图1A、图1B)后,与对照组比较,PIDC左心室组织中有137个DEGs,其中70个表达上调、67个表达下调。对DEGs绘制火山图进行可视化分析(见图1C)。取差异显著前100个基因绘制热图(见图1D)。

图1 PIDC基因差异表达分析(A为原始矩阵数据归一化处理前;B为原始矩阵数据归一化处理后;C为基因差异表达分析火山图;D为基因差异表达分析热图。其中红色表示上调;蓝色表示下调)
2.2 DEGs富集分析
GO功能富集分析显示:DEGs主要参与对外部刺激的正向调节、炎症反应的调节、中性粒细胞的激活和介导的免疫、细胞-基质黏附的正向调节、细胞外结构、含胶原的细胞外基质等。详见图2A。KEGG通路富集分析显示:DEGs参与细胞凋亡、胰岛素抵抗、花生四烯酸代谢和缺氧诱导因子1(hypoxia-inducible factor 1,HIF-1)信号通路等。详见图2B。

图2 GO功能富集和KEGG通路富集分析气泡图(A为GO富集分析;B为KEGG富集分析)
2.3 PPI网络分析及核心基因筛选
对DEGs进行PPI网络分析,将离散节点隐去后,构建的初始PPI网络中节点(基因/蛋白)135个、边(蛋白互作关系)186条,平均节点度值为2.76,PPI富集P值<1.0e-16。输出TSV格式文件并导入Cytoscape软件,以CytoHubba插件进行拓扑计算,取MCC排名居前5位的基因作为核心基因,即信号转导与转录激活因子3(signal transducer and activitor of transcription 3,STAT3)、CD68、高亲和力IgE受体γ亚基基因(high-affinity IgE receptor gamma subunit gene,FCER1G)、细胞周期蛋白D1(cyclinD1,CCND1)和CD163。亮色标记的节点为核心基因,颜色深浅代表MCC值大小,共36个节点、100个边,详见图3A。以分组比较呈现两组5个核心基因的差异表达,详见图3B。
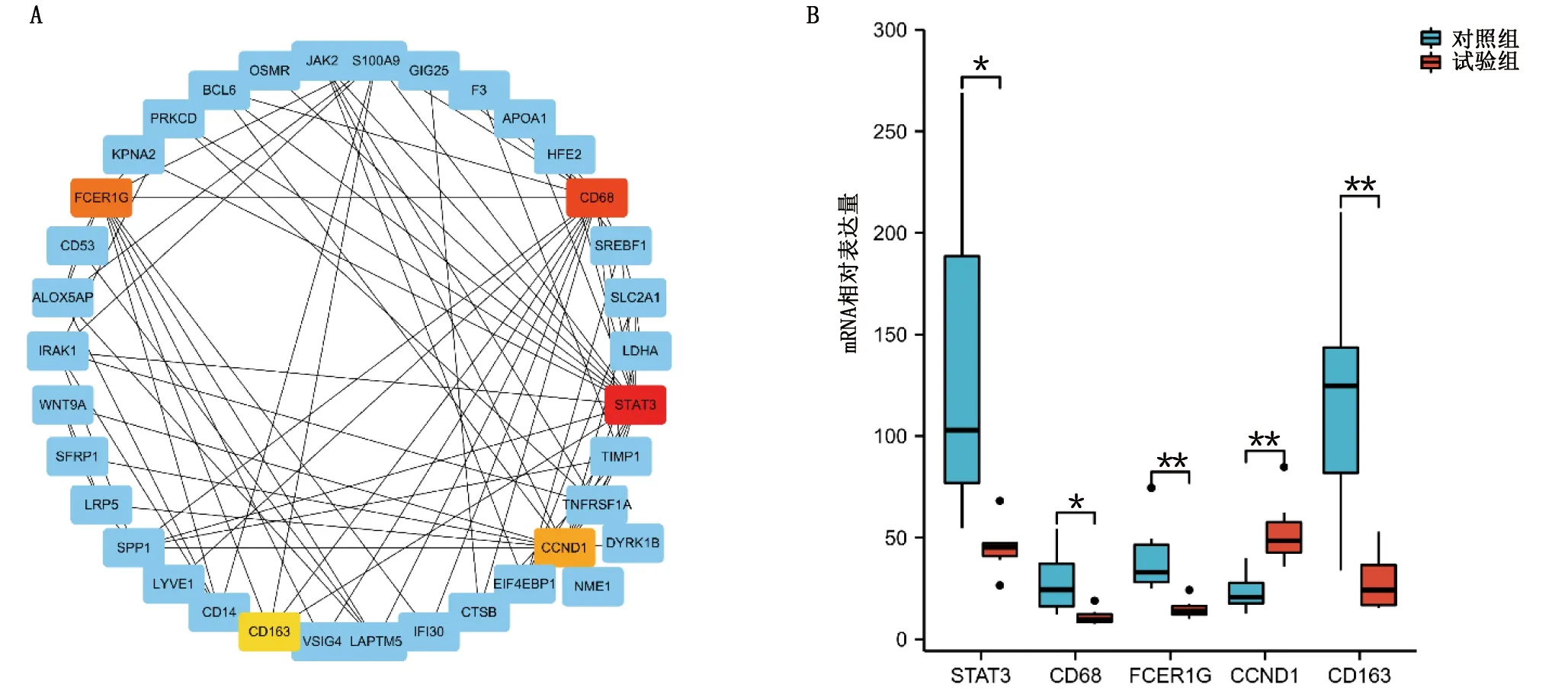
图3 差异基因构成的PPI网络中鉴定核心基因及分组比较(A为鉴定核心基因;B为分组比较柱状图。*P<0.05,**P<0.01。色彩鲜明的节点为5个核心基因)
2.4 核心基因与心肌病、心力衰竭的关系
核心基因与心肌病、心力衰竭的相互作用评分分析提示,5个核心基因STAT3、CD68、FCER1G、CCND1和CD163与心肌病、心力衰竭关联紧密。详见图4。可见核心基因与心肌病、心力衰竭关联性得分情况,得分越高提示关联性越大。
2.5 免疫细胞浸润分析
与对照组比较,试验组左心室组织中央记忆型CD8+T细胞含量较高,活化的树突样细胞、骨髓来源的抑制性细胞、自然杀伤T细胞、浆细胞样树突状细胞、滤泡辅助性T细胞含量较低,差异均有统计学意义(P<0.05)。详见图5、图6。图中红色表示上调,蓝色表示下调。

图5 PIDC免疫细胞浸润分析热图
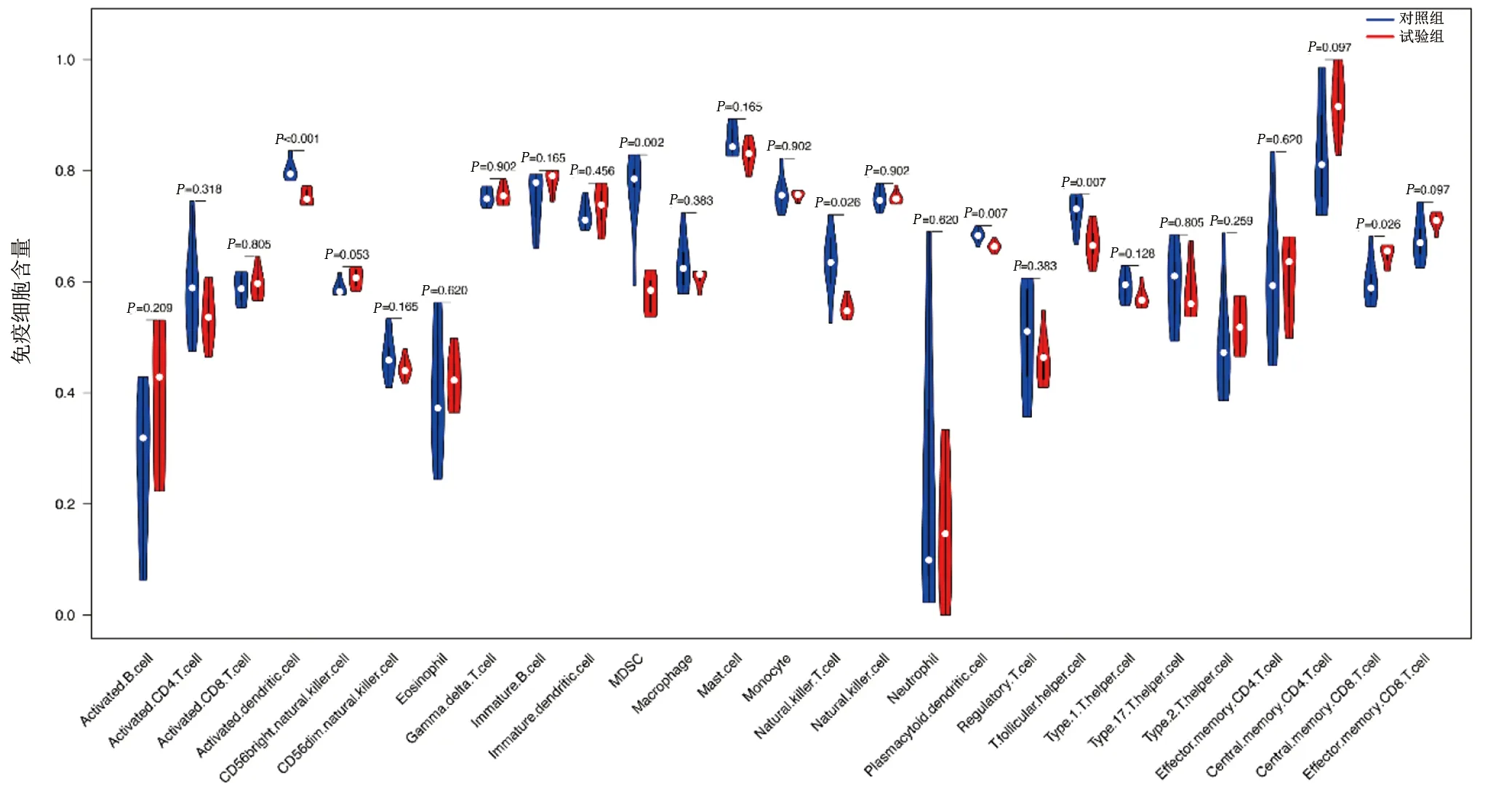
图6 PIDC免疫细胞浸润分析小提琴图
3 讨 论
PIDC具有较高的发病率、死亡率,是小儿心脏移植的主要原因之一,严重威胁儿童身心健康。该病发病机制复杂,尚未明确,因而治疗难度较大。本研究基于生物信息学分析,探讨免疫相关的分子机制,并筛选出核心基因及浸润的免疫细胞,从而为探讨疾病机制、寻求治疗靶点提供理论依据。
首先,本研究发现PIDC左心室组织中存在137个DEGs。其次,GO功能富集和KEGG通路富集分析发现DEGs的功能主要集中在对外部刺激的正向调节、炎症反应的调节、中性粒细胞的激活和介导的免疫、细胞-基质黏附的正向调节、细胞外结构、含胶原的细胞外基质等;这些差异基因主要富集在细胞凋亡、胰岛素抵抗、花生四烯酸代谢和HIF-1信号通路等。免疫介导的炎症损伤是DCM发生发展中的关键过程之一[14]。已有研究报道,炎症反应由凋亡分子Fas配体(Fas ligand,FasL)通过心肌细胞和成纤维细胞中的细胞外信号调节激酶1/2(extracellular-signal-regulated kinase 1/2,ERK1/2)被激活,最终诱导DCM和心力衰竭的发生。阻断ERK1/2通路、抑制炎症等可阻止疾病的进展[15]。有研究显示,DCM中炎症趋化因子水平显著升高[16]。中性粒细胞作为炎症反应的重要参与者,其水平的高低与DCM心力衰竭的严重程度相关[17]。心脏细胞外基质是一个动态分子网络,为心脏功能提供结构支撑。细胞-基质相互作用的改变、细胞外基质重塑被认为是心肌扩张过程中细胞重构的重要组成部分[18-19]。细胞外基质重塑通过影响细胞归巢、成纤维细胞激活、局部黏附蛋白表达参与DCM等,导致心力衰竭发生发展[20]。
同时本研究结果显示,细胞凋亡是PIDC发生发展过程中的关键致病因素之一。转录因子p53是人体重要的管家基因,可维持正常的心脏结构和生理功能。已有研究报道,p53水平升高促进DCM中的心脏重构,这可能是通过诱导细胞凋亡、细胞周期停滞实现的[21]。有研究显示,基因PPP1R13L双等位变异与PIDC强烈相关,而PPP1R13L的功能是编码p53蛋白的促凋亡蛋白抑制剂[22]。阿霉素作为一种化疗药物,可提高肿瘤病人生存率,但其诱导DCM的作用也有报道,认为这种不良反应是阿霉素通过增加线粒体膜通透性,进而增加心肌细胞凋亡实现的[23]。本研究结果显示,胰岛素抵抗在PIDC中有致病作用。胰岛素抵抗通过增加游离脂肪酸氧化,造成心肌细胞能量代谢障碍,促进DCM的形成;反之,DCM通过促炎细胞因子释放、儿茶酚胺等激素释放,交感神经系统的激活,进一步加重胰岛素抵抗[24]。上述研究均提示,干预细胞凋亡和胰岛素抵抗可能成为PIDC的治疗方向。有研究显示,花生四烯酸CYP450酶代谢的产物可能在克山病和DCM的发病机制中发挥着关键作用,如CYP2C19基因上调和蛋白表达频繁可能是一种保护性补偿反应,而CYP1A1可能加重心肌损伤[25]。本研究富集分析结果显示,差异基因显著参与花生四烯酸代谢通路,提示该通路可能作为PIDC心力衰竭治疗的靶点[26]。HIF-1是缺氧/缺血血管反应的主要调节因子,驱动数百个基因的转录激活,涉及血管反应、血管生成及骨髓源性血管生成细胞的动员和归巢[27]。特发性扩张型心肌病表现为血管生成缺陷,且心外膜血管疏松与HIF-1增加相关[28]。本研究发现多个基因以下调方式参与HIF-1信号通路中,提示该通路可能被抑制。干预HIF-1信号通路,可影响心肌血管生成,可能是PIDC的治疗靶点之一。
其次,本研究通过PPI网络分析筛选了5个与心肌病及心力衰竭关系密切的核心基因。STAT3是对心脏具有保护作用的转录因子,STAT3的激活通过增强心肌细胞的预适应和后适应、抗炎、诱导生存基因表达、维持线粒体功能等多种方式发挥心脏保护作用。既往研究显示,STAT3通路的下调可诱发DCM的心肌不良重构[29]。有研究表明,心肌细胞特异性STAT3通过维持肌萎缩蛋白和α-肌聚糖的表达水平,防止扩张表型的发生和发展[30]。本研究发现STAT3的水平在PIDC中是显著下调的。CD68是巨噬细胞的标志,目前关于其在心肌病和心力衰竭中作用的研究结果存在争议。有研究显示,巨噬细胞作为炎症过程的重要参与者,其在慢性炎症的心力衰竭病人中水平显著升高[31]。相关研究显示,与健康对照组相比,DCM病人心肌细胞中的巨噬细胞数量无显著变化[32-33]。CD163是M2型巨噬细胞的标志,其在DCM中的研究结果存在争议。有研究报道,CD163阳性细胞数量的增加与DCM病人较差的预后显著相关,是心肌组织中胶原含量的独立预测因素,可能参与DCM的心室重塑[34]。有研究显示,CD163水平与DCM的严重程度无关[35]。本研究分析发现CD68和CD163的水平在PIDC病人中均是降低的。这些结果提示巨噬细胞可能通过复杂的机制参与PIDC的发生发展,仍需进一步研究。FCER1G作为固有免疫基因,参与炎症通路,诱导多种疾病发生[36]。一项体细胞和单细胞RNA测序数据的综合分析发现,FCER1G参与DCM的免疫过程[37],其与DCM或心力衰竭的关系,在这2种疾病病理生理状态中的作用仍待进一步研究。CCND1是重要的细胞周期调节蛋白,促进细胞周期从DNA合成前期(G1期)到DNA复制期(S期)的转换,其水平的上调参与多种肿瘤的进展[38]。基因肌联蛋白(TTN)的功能是编码肌节中最大的结构蛋白,其截段突变导致TTN缺乏是家族性和散发性DCM的主要原因。Liao等[39]研究显示,CCND1上调可抑制TTN缺乏引起的DCM,结果提示TTN不足导致DCM促进心肌细胞的细胞周期治疗,CCND1是其中一个重要的治疗靶点。核心基因在PIDC的发病机制仍待研究,可能成为治疗PIDC和DCM的新靶点。
最后,本研究进行免疫细胞浸润分析发现,与对照组比较,试验组6种细胞含量有显著差异。CD8+T细胞在PIDC的心肌组织中含量升高。Luppi等[40-41]研究显示,DCM病人心肌组织中有CD8+T淋巴细胞浸润。Rao等[42]进一步研究提示,增多的CD8+T细胞主要位于纤维化的心肌中。上述研究结果提示CD8+T细胞在PIDC的发病机制,特别在心室重构中发挥着重要的致病作用。DCM和心力衰竭病人中树突样细胞减少[32,43]。本研究分析发现树突样细胞在PIDC的心肌组织中含量降低。关于骨髓来源的抑制性细胞与DCM的研究较少,Zhang等[44]研究显示,DCM病人循环中数量显著升高,且与N末端脑钠肽前体呈正相关,与左室射血分数呈负相关。本研究分析发现,骨髓来源的抑制性细胞在PIDC病人心肌组织中减少,不一致的研究结果提示其在DCM发生发展中的作用有待进一步研究。本研究发现,PIDC心肌组织中自然杀伤T细胞含量减少。有研究显示,自然杀伤T细胞在DCM中含量降低[45]。已有基础研究显示,DCM小鼠脾脏中自然杀伤T细胞数量减少,补充外源性的树突样细胞可激活体内的自然杀伤T细胞,延长DCM小鼠生存,预防心脏收缩功能下降,抑制间质纤维化[46]。这些结果提示免疫细胞调节治疗可能成为治疗DCM的新策略。
综上所述,炎症及免疫反应在PIDC的发病机制中发挥着重要作用,5个核心基因和6种免疫细胞与PIDC发生发展密切相关,是其防治的潜在靶点。本研究对PIDC关键基因及信号通路进行了分子机制探讨,并辅以免疫细胞浸润分析,后期仍需进一步深入探讨免疫浸润细胞与关键基因或通路的关联性。
猜你喜欢
中国临床医学影像杂志(2019年1期)2019-04-25
中国中医药信息杂志(2016年5期)2016-12-01
中西医结合心脑血管病杂志(2016年20期)2016-03-01
山东医药(2015年14期)2016-01-12
中国病理生理杂志(2015年8期)2015-12-21
医学研究杂志(2015年3期)2015-06-10
国际心血管病杂志(2015年5期)2015-02-27
创业家(2015年1期)2015-02-27
遗传(2014年2期)2014-02-28
长江大学学报(自科版)(2014年27期)2014-02-27
