
高效液相色谱-高分辨轨道阱质谱联用法对兰索拉唑肠溶制剂的杂质谱研究
2023-11-06 01:39贾欢欢袁耀佐陈民辉张锦琳
中国药科大学学报 2023年5期
李 岩,贾欢欢,黄 青,袁耀佐,陈民辉,张锦琳*
(1江苏省食品药品监督检验研究院国家药品监督管理局化学药品杂质谱研究重点实验室,南京 210019;2中国药科大学药学院,南京 211198)
兰索拉唑属苯并咪唑类,是继奥美拉唑之后的新一代质子泵抑制剂[1],由日本武田制药株式会社研制,于1991 年在法国首次以商品名Takepron上市,1995 年通过美国FDA 认证[2]。主要作用于H+-K+-ATP 酶来发挥抑酸作用,临床广泛用于治疗各种消化功能紊乱性的疾病,如胃溃疡、十二指肠溃疡、反流性食管炎、吻合口溃疡、消化道出血、胃泌素瘤,以及其他症状较重或经 H2受体拮抗治疗无效的难治性溃疡等[3],对幽门螺杆菌也有抑制作用[4]。兰索拉唑遇酸、热、氧化等条件均易变质降解,因此对其有关物质的控制与研究尤为重要,因兰索拉唑酸不稳定的特点[5],原研剂型为口崩片和肠溶胶囊,国内市场上的口服剂型主要为肠溶片和肠溶胶囊,国产兰索拉唑肠溶片为改剂型品种,其杂质谱与原研制剂是否存在差异也是本研究的关注点之一。天津武田药品有限公司生产的兰索拉唑肠溶胶囊属于原研地产化品种,是国家局公布的仿制药参比制剂,因此本研究以其作为参比制剂进行相关研究。
英国药典、美国药典和中国药典均采用高效液相色谱法检查兰索拉唑的有关物质[6-8]。英国药典给出了6 个已知杂质,分别为杂质A、B、C、D、E和F;美国药典给出了3个已知杂质,为兰索拉唑N氧化物、杂质A 和杂质B(兰索拉唑硫醚),分别对应英国药典的杂质A、B和C;两者均对已知杂质进行了限度控制。但中国药典未给出已知杂质限度要求,仅对单个杂质和各杂质峰面积的和进行控制。目前中国食品药品检定研究院已有兰索拉唑杂质Ⅰ、Ⅱ、Ⅲ和Ⅳ对照品,分别对应英国药典的杂质A、B、D 和E。本研究首先利用二维在线除盐技术,采用高效液相色谱-高分辨轨道阱质谱联用检测方法,对兰索拉唑肠溶制剂在中国药典有关物质项下的检出杂质进行了结构推定,由于该色谱条件采用的流动相为甲醇-水-三乙胺-磷酸,与质谱检测器不兼容,且前期实验发现该方法中有部分杂质与辅料峰共同流出,为了进一步研究相关杂质,本研究又开发了兼容质谱检测器的有关物质色谱条件,在美国药典的兰索拉唑有关物质方法的基础上,尝试以兼容质谱检测器的乙酸铵替代三乙胺,冰乙酸替代磷酸,同时将流动相pH降低至6.5;为了延长杂质峰的保留时间,避免在切除溶剂峰时误将杂质峰一并切去,又优化了梯度洗脱程序,进而对新检出的杂质进行结构推定,解决了色谱流动相不适宜直接用于质谱分析的难题,又保证了不同来源的制剂中最大程度地检出有关物质,并考察了不同企业间产品杂质谱的差异性。
1 材 料
1.1 试 剂
兰索拉唑肠溶片(抽样样品,企业a ~ z),兰索拉唑肠溶胶囊(参比制剂,天津武田药品有限公司);兰索拉唑(批号100709-201705,含量99.6%);杂质Ⅰ(批号510046-201401);杂质Ⅱ(批号510047-201401);杂质Ⅲ(批号510048-201401);杂质Ⅳ(批号510049-201401)均来自中国食品药品检定研究院;杂质C(批号:S17041902,纯度99.94%)和杂质F(批号:S17110601,纯度99.3%)均由江苏奥赛康药业股份有限公司提供。乙腈、甲醇、甲酸、三乙胺为色谱纯,其他试剂均为市售分析纯,水为Milli-Q超纯水。
1.2 仪 器
LC-20AB 高效液相色谱仪(日本岛津公司);Ultimate 3000 -Q Exactive Orbitrap 液质联用仪(美国赛默飞世尔公司)。
2 方 法
2.1 二维在线脱盐液质联用方法
2.1.1 一维色谱条件 采用《中华人民共和国药典》有关物质的色谱条件[8]。色谱柱:Agilent Zorbax SB C18(4.6 mm × 250 mm,5 µm);流动相:甲醇-水-三乙胺-磷酸(600∶400∶5∶1.5)[用磷酸溶液(1→10)调节pH 至7.3],等度洗脱;流速:0.6 mL/min;柱温:30 ℃;进样体积:10 µL;检测波长:284 nm。
2.1.2 二维色谱条件 色谱柱:Waters C18T3(2.1 mm × 100 mm,1.7 µm);流动相A 为0.1%甲酸水溶液,流动相B 为0.1%甲酸乙腈溶液,梯度洗脱:0 ~ 5 min, 5% B;5 ~ 50 min, 5% B→95% B;50 min ~ 50.1 min, 95% B→5% B;50.1 ~ 60 min, 5% B。流速:0.3 mL/min;柱温:30 ℃;进样体积:10 µL;检测波长:284 nm。
2.1.3 质谱条件 离子源:ESI 源,正离子模式;扫描范围:一级质谱m/z100 ~ 1 000,二级质谱m/z50 ~ 1 000;鞘气流约4.58 L/min(40 arb),辅助气流约7.97 L/min(10 arb),喷雾电压:350 kV,毛细管温度:325 ℃,S-lens RF level:55.0,辅助器加热温度:350 ℃。
2.2 兼容质谱检测器的液质联用方法
2.2.1 色谱条件 色谱柱:Agilent Extend C18(4.6 mm × 150 mm,5 µm);流动相A 为25 mmol/L乙酸铵溶液,流动相B 为乙腈-25 mmol/L 乙酸铵溶液(4∶1)[用冰乙酸调节pH 至6.5];梯度洗脱:0 ~2 min, 10% B;2 ~ 40 min, 10% B→80% B;40 min ~50 min, 80% B;50 ~ 51 min, 80% B→10% B;51 min ~ 60 min, 10% B。流速:0.8 mL/min;柱温:25 ℃;进样体积:20 µL;检测波长:285 nm;
2.2.2 质谱条件 采用“2.1.3”项的质谱条件。校准方法:自动调谐优化电压,外标法校准质量数,子离子扫描模式为Fullms-dd/ms2。
2.3 溶液的制备
系统适用性溶液的制备:取兰索拉唑、杂质A、杂质B、杂质C、杂质D、杂质E、杂质F 对照品各适量,用甲醇-0.1 mol/L NaOH(1∶3)溶液溶解并稀释制成每毫升含兰索拉唑2 mg、杂质A、B、C、D、E、F各10 µg/mL的混合溶液。
供试品溶液的制备:取兰索拉唑肠溶片除去肠溶衣或肠溶胶囊内容物,研细,精密称取适量,加甲醇-0.1 mol/L NaOH(1∶3)溶液制成每毫升含兰索拉唑2 mg的溶液,滤过,取续滤液作为供试品溶液。
对照溶液:精密量取供试品溶液1 mL,至100 mL 量瓶中,用甲醇-0.1 mol/L NaOH(1∶3)稀释至刻度,摇匀,作为对照溶液。
3 结 果
3.1 二维在线除盐液质联用法检出杂质的结构推定
前期实验中,本研究对抽样获得的19 家企业生产的兰索拉唑肠溶片和8 家企业生产的兰索拉唑肠溶胶囊按照中国药典方法进行了有关物质检查,选出了5 批杂质较多的样品,均为肠溶片,与1 批参比制剂一起采用“2.1”项方法,利用二维在线除盐技术对检出杂质进行结构推定。按照中国药典规定相对保留时间0.25之前的色谱峰忽略不计,通过对照品定位和质谱信息发现,杂质D 和杂质E在紫外图谱中无法分离,但质谱提取离子流图可以实现有效分离,分别标记为杂质1和杂质2,其余杂质按出峰顺序依次编号,图1为检出杂质最多的2批样品,覆盖了全部9个杂质。

Figure 1 HPLC-UV chromatogramphys of representative samples of lansoprazole enteric formulation
通过与已知杂质的保留时间和质谱数据比较,归属了5 个色谱峰为已有对照品的已知杂质,分别为杂质D、E、A、B、C,在图中色谱峰编号依次为1、2、6、7、9,其余4 个杂质为未知杂质。为了更好地对杂质结构进行分析,首先对兰索拉唑本身结构,其一级、二级质谱行为,主要二级碎片结构进行全面解析,结果见图2;其中分子离子峰为m/z370.083 2,m/z252.029 7,m/z119.060 5 为其典型特征碎片,m/z119.060 5 可能为苯并咪唑碎片离子,m/z252.029 7可能为兰索拉唑脱去苯并咪唑的碎片离子。通过252、119、136等特征差异碎片的比对,可从质谱角度推测出未知杂质结构变化位点。

Figure 2 Structural analysis of major fragments by lansoprazole secondary mass spectrometry
以杂质4 为例,提取杂质4 色谱峰顶点的质谱图,见图3,其中主要离子为370.083 5,实际测定质荷比与推测分子式理论质荷比一致(diff =0.8 × 10-6),推测化合物分子式为C16H14F3N3O2S。结合杂质4的二级质谱信息,推测其化学结构式见表1。以上述推测的杂质4结构式进行质谱碎片断裂机制的推导,该化合物为兰索拉唑的同分异构体,二级质谱碎片与兰索拉唑高度吻合,变化位点有多个可能性,目前信息不足以证明。
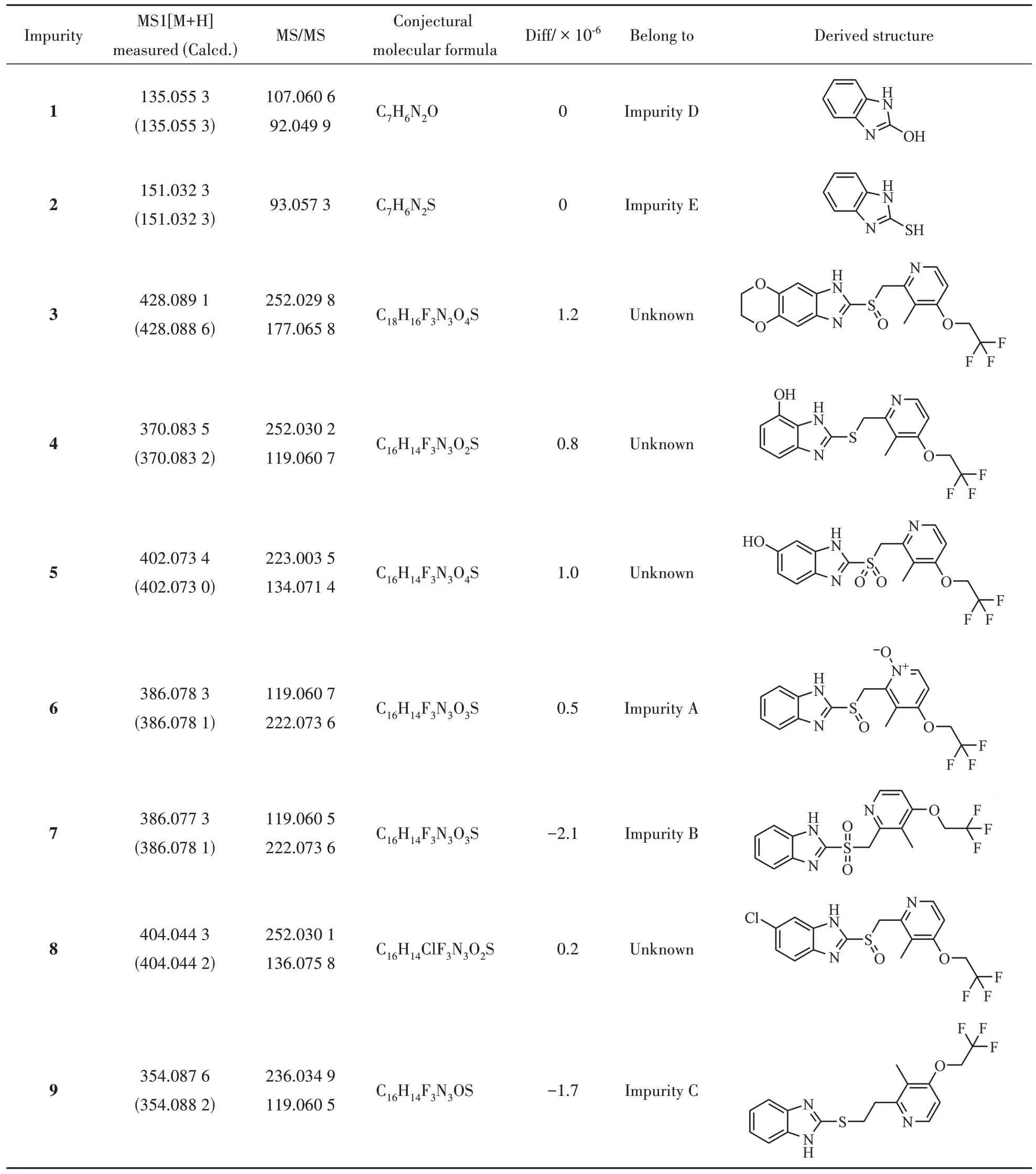
Table 1 Lansoprazole main impurities mass spectrometric information
Figure 3 Mass spectrum of impurity 4
用上述方法结合碳氢比和二级质谱信息分别对其余未知杂质进行分析,推测杂质3 分子式为C18H16F3N3O4S,与兰索拉唑相比,分子式中多了两个C,两个H 和两个O,初步推测为兰索拉唑加环产物,二级碎片m/z252.029 8 与兰索拉唑一致,根据碎片m/z177.065 8 推测加环位点为苯并咪唑;推测杂质5 分子式为C16H14F3N3O4S,与兰索拉唑相比,分子式中多了两个O,二级碎片m/z223.003 5和m/z134.071 4 与兰索拉唑均不一致,推测兰索拉唑结构式两端均有变化位点,仍需进一步的研究手段证明;推测杂质8分子式为C16H14ClF3N3O2S,与兰索拉唑相比,分子式中多了1 个Cl,主要二级碎片m/z252.030 1、m/z136.075 8 与兰索拉唑一致,推测变化位点为苯并咪唑。其MS 数据、分子式和推测所得结构式分别如表1 所示。其中杂质1、2、6、7、9 分别为已知杂质D、E、A、B、C,杂质3、4、5、8为本研究中新发现的杂质。
3.2 兼容质谱检测器的液质联用法检出杂质的结构推定
前期实验发现,在《中华人民共和国药典》方法中,有部分杂质的相对保留时间在0.25之前,与辅料峰共同流出,为了进一步研究兰索拉唑相关杂质,本实验建立了兼容质谱检测器的有关物质测定方法,对新检出的杂质结构进行推定,考察不同企业产品间杂质谱的差异性。该方法与二维在线脱盐方法相比,保证了不同来源原料及制剂中最大程度地检出杂质。
采用“2.2”项方法对5 批代表性样品和1 批参比制剂进行研究,紫外典型色谱图见图4,企业c和企业b 的样品杂质较多,共检出14 个杂质,包括了本研究中的主要杂质,对其进行编号(共编号1 ~14)。其中峰1 ~ 峰9 与二维在线除盐质谱分析结果一致,在“3.1”项中已对其进行推定,另有5 个(峰10 ~ 峰14)是本实验新检出的未知杂质。根据精确相对分子质量和碎片裂解规律进行推测,可能结构结果及质谱信息见表2。
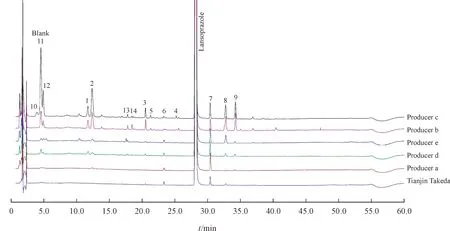
Figure 4 HPLC-UV one-dimensional chromatographic analysis of the samples
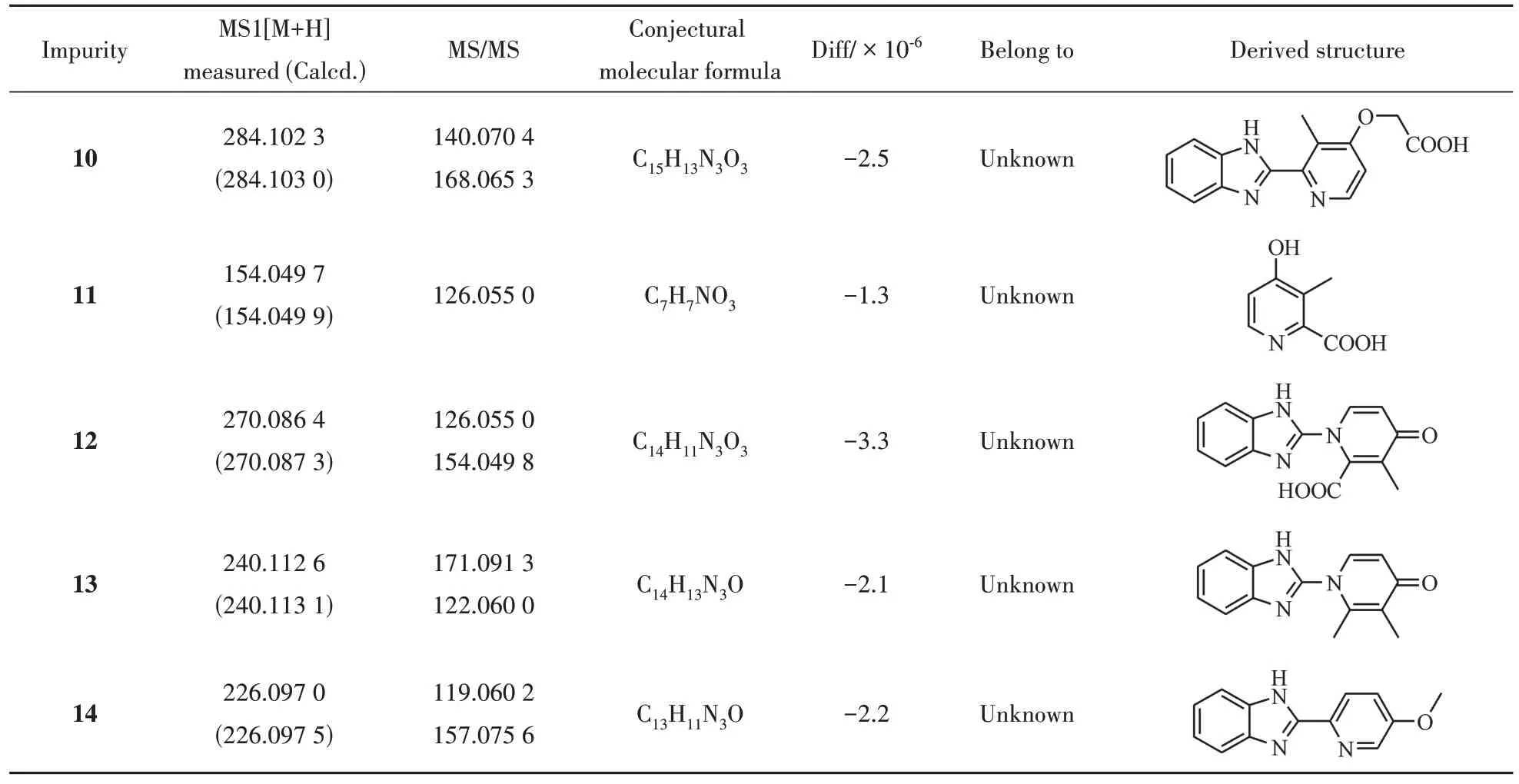
Table 2 Lansoprazole main impurities mass spectrometric information under mass spectrometric compatible conditions
以杂质10 为例,提取杂质10 色谱峰顶点的质谱图,如图5所示,其中主要离子是m/z284.102 3。实际测定质荷比与推测分子式理论质荷比一致(diff = -2.5 × 10-6);故推测得化合物分子式为C15H13N3O3,推测可能为兰索拉唑脱去亚磺酰基。进一步结合杂质10 的二级质谱信息,推测其化学结构式见表2。用上述方法分别对未知杂质11 ~14 进行分析,其MS 数据、分子式和推测所得结构式分别如表2 所示,推测杂质11 分子式为C7H7NO3,其二级碎片离子较少,结构相对稳定;杂质12 与杂质11 具有相同的碎片离子,故在推测的杂质11结构上对杂质12进行推定。
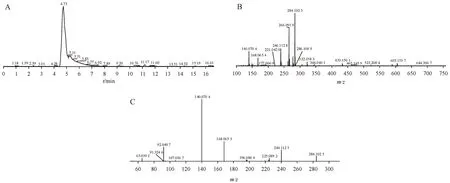
Figure 5 Mass spectrum of impurity 10
3.3 杂质来源分析
通过酸降解(0.1 mol/L盐酸溶液,5 min)、碱降解(1 mol/L氢氧化钠溶液,2 h)、热降解(90 ℃,2 h)、氧化降解(3%双氧水,30 min)、光降解(254 nm 紫外光,24 h)的强制降解实验结果(详见图6)、兰索拉唑原料药的合成工艺以及文献资料[9-12],对杂质来源进行分析,推测杂质D、未知杂质3、未知杂质4、未知杂质5、杂质A、杂质B、未知杂质8、未知杂质10、未知杂质11、未知杂质12、未知杂质13 为降解杂质,杂质E和杂质C可能为合成原料药合成工艺中的副产物。
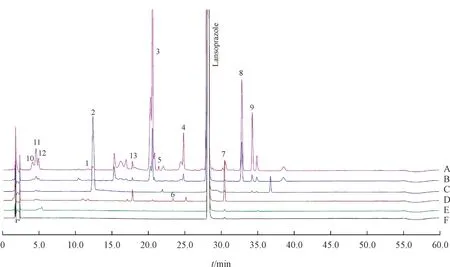
Figure 6 Chromatogram of forced degradation of lansoprazole raw material
3.4 样品测定
对抽样获得的20家企业生产的兰索拉唑肠溶片和8 家企业生产的兰索拉唑肠溶胶囊采用USP42 兰索拉唑肠溶胶囊的有关物质方法进行HPLC-UV 测定,按加校正因子的自身对照法计算,小于0.025%的色谱峰忽略不计。各批次样品测定结果见表3。结果可见,所有样品均检出杂质B,不同生产企业的产品杂质谱存在明显差异,参比制剂的杂质总量较低,国产兰索拉唑肠溶片之间的杂质种类和含量都有较大差距。

Table 3 Summary table of the related substance results of lansoprazole enteric formulation
4 讨 论
对于兰索拉唑肠溶制剂有关物质的检查,中国药典的色谱条件无法使杂质D 和杂质E 完全分离,无法准确分别测定其含量;而美国药典的色谱条件虽然分析时间长,但能对6个已知杂质实现有效分离。因此基于美国药典色谱条件开发的兼容质谱的色谱条件无论在检出杂质个数还是总量都优于中国药典方法,尤其在杂质较多的样品中,可以分离出更多杂质。
本文采用了两种液质联用方法对兰索拉唑肠溶片和肠溶胶囊的杂质谱进行研究,在二维在线除盐方法上,推定出9 个杂质的结构,其中5 个为已知杂质(分别对应英国药典杂质D、杂质E、杂质A、杂质B 和杂质C),4 个为新鉴定的未知杂质,该方法可以对《中华人民共和国药典》条件下检出杂质进行结构推定;采用新建立的质谱兼容流动相色谱条件串联高分辨质谱直接进样分析,共检测出14 个有关物质,其中5 个为已知杂质,4 个未知杂质与二维在线脱盐方法结果一致,另外5个为该条件下新检出的杂质。对主成分兰索拉唑的一级和二级质谱图的碎片结构解析中发现,兰索拉唑的分子离子峰是m/z370.083 2,其中m/z252.029 7,m/z119.060 5为其典型特征碎片,m/z252.029 7可能是兰索拉唑脱去苯并咪唑的碎片离子峰,m/z119.060 5 可能是兰索拉唑结构中的亚硫酰基与碳相连单键断裂形成的苯并咪唑的碎片离子。m/z252.029 82碎片离子和分子离子峰为m/z234.019 5相差18,失去一分子水,推测可能是亚硫酰基发生断裂。本文在此基础上对未知杂质结构进行初步推定,但还需要借助NMR,IR 等手段进行进一步研究。
国产兰索拉唑肠溶片为改剂型品种,不同产品间的杂质个数和含量都存在较大差异,可能与原料药来源和生产工艺不同相关。本研究对兰索拉唑肠溶制剂杂质谱进行研究,对未知杂质的结构以质谱的碎片信息进行了推测和来源归属,于兰索拉唑肠溶制剂的质量控制和工艺评价具有指导意义。
猜你喜欢
中学生数理化·自主招生(2023年2期)2023-08-22
中国典型病例大全(2022年13期)2022-05-10
中华养生保健(2020年8期)2021-01-14
中学生数理化·高一版(2020年6期)2020-07-25
中华胃食管反流病电子杂志(2017年2期)2017-10-27
家庭用药(2017年5期)2017-05-26
中学生数理化·高三版(2016年12期)2017-03-02
西南军医(2016年5期)2016-01-23
中国卫生标准管理(2015年25期)2016-01-14
中国药业(2014年17期)2014-05-26
