
CFTR基因突变致囊性纤维化家系报道及文献复习
2023-11-03 04:45王丽刘瑞寒李秋波辛美云
中国中西医结合儿科学 2023年5期
王丽, 刘瑞寒, 李秋波, 辛美云
囊性纤维化是一种累及多系统多脏器的常染色体隐性遗传病,由囊性纤维化跨膜传导调节因子(cystic fibrosis transmembrane conductance regulator,CFTR)基因突变所致,主要表现为慢性进行性阻塞性肺部疾病,并伴有其他多系统多脏器受累,如营养不良、吸收不良、囊性纤维化相关的糖尿病、肝脏疾病等。其中肺部病变是其主要表现,也是其主要的致死原因,几乎所有的患儿均会出现慢性阻塞性肺病合并感染,导致肺功能进行性丧失。Davis[1]于1938年首次报道该病,囊性纤维化在欧美白种人群中发病率约为1∶2 500,而亚洲人种中该疾病少见,日本发病率约为1∶350 000。由于我国主要是黄种人,发病率较低,尚无发病情况的流行病学统计数据[2]。但随着近年来基因检测技术的发展以及对该病的认识逐渐提高,我国对囊性纤维化的诊断也逐年增多。我们将兄妹2人同患囊性纤维化的临床诊疗经过及基因检测分析报告如下,以提高临床医生对该病的认识。
1 临床资料
例1,患儿女,5岁7个月,因“咳嗽5年半,反复腹痛半年,加重4 d”入院。晨起咳嗽、经常咳痰,为绿色痰液,曾多次诊断“慢性肺炎”,给予口服及静脉滴注抗生素治疗可好转,仍每日均有咳嗽,活动耐受性差;近半年出现腹痛,阵发性,脐周为著,无规律性,可自行缓解,无呕吐,无多饮、多尿。个人史:系G2P2,足月顺产娩出,出生体质量2.75 kg,无窒息、缺氧史,10个月时会叫“爸爸、妈妈”,1岁2个月会走,智力正常。既往史:有5次“脱肛”史,有1次需用手还纳,余可自行还纳;自3岁起进食油性食物后出现脂肪泻。家族史:父母非近亲婚配,均体健,有一哥哥。
体格检查:体温36.9 ℃,脉搏127次/分,呼吸40次/分,体质量14.0 kg,身高108 cm。精神差,营养不良,气促。皮下脂肪薄,弹性欠佳。口唇尚红润。桶状胸。三凹征阳性。双肺呼吸音粗,可闻及固定干啰音。心率127次/分,心音有力,律齐。腹部膨隆,腹肌紧张,叩诊鼓音,脐周压痛,肝脾触诊不理想。肢端暖,毛细血管再充盈时间小于2 s,杵状指/趾。
辅助检查:血常规:白细胞计数17.2×109/L,中性粒细胞74.6%,嗜酸粒细胞0.24%,血小板计数414×109/L,血红蛋白138 g/L,C反应蛋白11.4 mg/L。粪便常规脂肪球++。肾功能、心肌酶、血淀粉酶、电解质、血脂正常。肝功能:丙氨酸氨基转移酶189.9 U/L,天门冬氨酸氨基转移酶181 U/L。糖化血红蛋白8.4%。胰岛素、C肽正常范围。糖尿病相关抗体检测阴性。1-3-β-D葡聚糖18.412 ng/L(正常值0~159 ng/L),曲霉菌血清学试验0.84(≥0.5为阳性)。免疫球蛋白11 900 IU/mL(参考值≤90 IU/mL)。免疫球蛋白定量:IgM 2.32 g/L(参考值0.4~2.3 g/L),IgG 20.8 g/L(参考值7~16 g/L),IgA 4.74 g/L(参考值0.7~4 g/L)。T淋巴细胞亚群大致正常。支气管灌洗液培养提示铜绿假单胞菌。心脏彩超、门静脉彩超:正常。胸部CT示:双肺见多发散在斑片状及粟粒状高密度影,边缘模糊。双肺支气管管壁增厚,管腔略狭窄,部分欠通畅并见高密度影充填;双肺见多发囊状、柱状扩张支气管影(图1)。腹部CT示:(1)肝硬化;肝脏多发弥漫性低密度影。(2)胰腺体积小并脂肪浸润。(3)胆囊移位并胆汁淤积。(4)结肠及小肠明显扩张、积气、内容物潴留并管壁增厚、肿胀(图2)。副鼻窦CT示:双侧上颌窦及筛窦炎,双下鼻甲略肥厚(图3)。
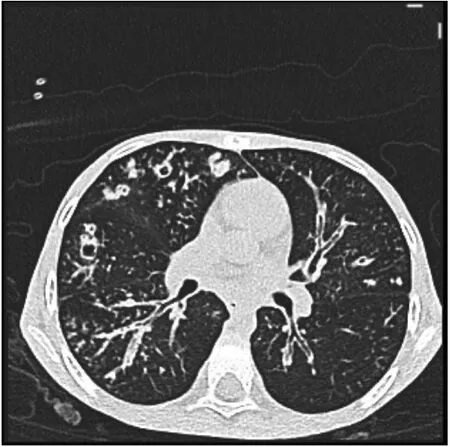
注:双肺下叶及左肺上叶支气管壁增厚、管腔狭窄并柱状及串珠状改变、右肺上叶及中叶支气管壁增厚并囊状扩张,部分扩张支气管内黏液栓阻塞
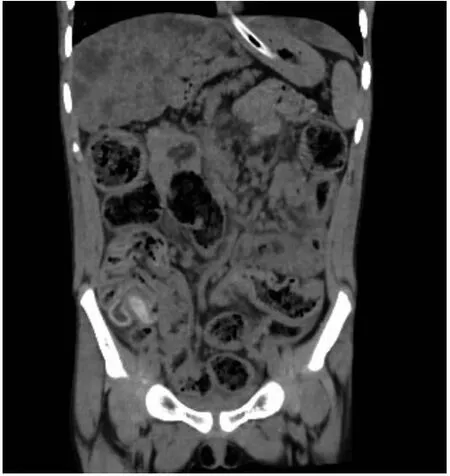
注:肝脏边缘呈波浪状及扇贝状改变,肝内多发斑片及结节状密度减低影,胰腺体积明显减小、萎缩并实质脂肪样变性-呈羽毛状改变;结肠及小肠明显扩张/积气、内容物潴留并管壁增厚、肿胀
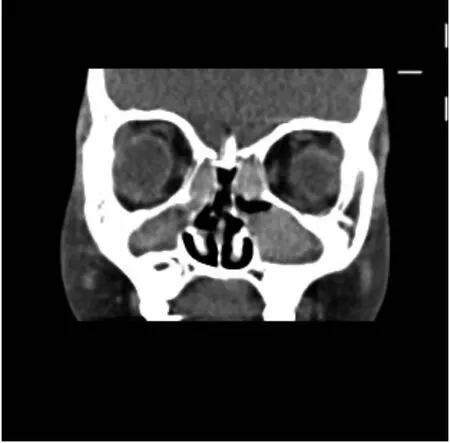
注:双侧筛窦、上颌窦及鼻腔黏膜增厚、密度增高,上颌窦及鼻腔内囊肿形成:鼻炎、副鼻窦炎
例2,患儿男(例1哥哥),8岁,因“反复咳嗽8年”入院。曾多次诊断“慢性肺炎、支气管扩张症”,可咳出黄绿色痰液,多次住院静脉滴注抗生素及行纤维支气管镜肺泡灌洗治疗,症状好转,但仍有咳嗽。个人史:系G1P1,足月顺产娩出,出生体质量3.05 kg,无窒息、缺氧史。3个月会抬头,7个月会坐,10个月会喊“爸爸、妈妈”,1岁2个月会走。既往史:无特殊。家族史:父母非近亲婚配,均体健,有一妹妹,病史如上述。
体格检查:体温36.5 ℃,脉搏120次/分,呼吸32次/分,体质量15.8 kg,身高115 cm。神志清,精神尚可,营养差。呼吸促,面色暗黄,皮肤黏膜黝黑。咽部充血,双侧扁桃体Ⅰ度肿大。三凹征阳性,双肺呼吸音偏低,可闻及较多痰鸣音、喘鸣音,密集中小水泡音。心音有力,律齐。腹软,肝肋下8.0 cm,剑突下9.5 cm,质韧,脾肋下未触及,未触及包块。杵状指/趾。
辅助检查:血常规:白细胞计数18.3×109/L,中性粒细胞73%,嗜酸粒细胞0.5%,血红蛋白149 g/L,血小板计数414×109/L,C反应蛋白8 mg/L。粪便常规脂肪球++。肾功能、心肌酶、血淀粉酶、电解质、血脂正常。肝功能:丙氨酸氨基转移酶85.3 U/L,天门冬氨酸氨基转移酶52 U/L。糖化血红蛋白6.2%。IgE 36.6 IU/mL(参考值≤90 IU/mL)。1-3-β-D葡聚糖<10 ng/L(正常值0~159 ng/L),曲霉菌血清学试验0.32(≥0.5为阳性)。免疫球蛋白定量:IgM 1.89 g/L(参考值0.4~2.3 g/L),IgG 17.0 g/L(参考值7~16 g/L),IgA 3.69 g/L(参考值0.7~4 g/L)。T淋巴细胞亚群大致正常。支气管灌洗液培养提示铜绿假单胞菌。心脏彩超、门静脉彩超:正常。胸部CT示:(1)双肺多发炎症。(2)双肺多发局部支气管扩张并感染,部分黏液栓形成(图4)。腹部CT示:(1)不均匀脂肪肝。(2)胰腺萎缩、脂肪化(图5)。副鼻窦CT示:(1)全组副鼻窦炎。(2)鼻中隔左偏(图6)。

注:左肺上叶舌段及下叶背段、右肺上叶前段及下叶背段支气管壁增厚、管腔柱状扩张/局部伴狭窄、部分呈轨道征,左肺下叶背段局部呈串珠状改变,右肺上叶前段支气管黏液栓阻塞-管腔不通畅
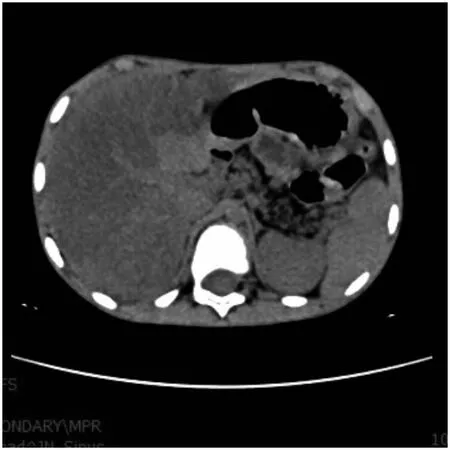
注:肝脏密度弥漫性减低,明显低于同层脾实质密度。胰腺体积萎缩、并实质脂肪样变性
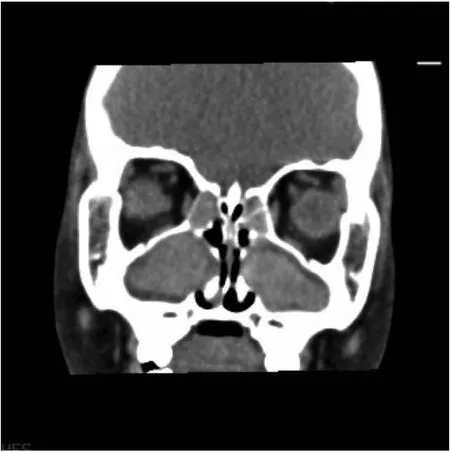
注:双侧筛窦、上颌窦及鼻腔黏膜增厚,鼻炎、副鼻窦炎症(上颌窦及筛窦内密度增高,合并鼻窦囊肿形成)
诊断及治疗:两例患者均给予加强呼吸道管理、体位引流、多次支气管肺泡灌洗、特布他林、布地奈德、乙酰半胱氨酸雾化吸入、头孢哌酮舒巴坦钠抗感染,例1加用伏立康唑抗真菌等治疗,均咳嗽减轻、复查肺部CT好转出院。出院时抽取例1外周静脉血行全外显子高通量测序,例2及其父母外周静脉血行Sanger验证。结果显示:例1CFTR基因有1个杂合缺失和1个半合子突变:exon18-20杂合缺失;在3196号核苷酸由胞嘧啶C变为胸腺嘧啶T(c.3196C>T)的半合子突变,导致第1066号氨基酸由精氨酸变为半胱氨酸(p.R1066C),为错义突变。根据美国医学遗传学与基因组学学会指南,该2个变异均判定为致病性变异。通过Sanger验证,例2及其母亲均检测到CFTR基因杂合变异:c.3196C>T(p.R1066C),其父亲该位点无变异,但临床表现例2符合囊性纤维化,我们推测例2另外一条染色体可能与其妹妹一样为杂合缺失,故对其父亲及例2再进行多重连接依赖式探针扩增技术(multiplex ligation-dependent probe amplification,MLPA),检测到其父亲及例2均存在CFTR基因外显子18-20杂合缺失突变。最终兄妹二人均确诊囊性纤维化,其父母均为囊性纤维化携带者。
2 讨论
囊性纤维化是白种人中最常见的致命性遗传病,可累及多系统多器官的外分泌腺。该病是由于CFTR基因突变所致,该基因最早于1989年由Riordan等[3]克隆分离成功。该基因是一种环磷酸腺苷(cAMP)依赖性调节氯离子通道蛋白,位于7号染色体长臂,编码1 480个氨基酸,通过氯化物途径调节上皮细胞的功能。基因突变可导致其编码的蛋白活性下降,使外分泌液中Na+、Cl-浓度异常增高,导致气道、肠道、汗腺等分泌物脱水。如果分泌物滞留并阻塞气道,则导致慢性阻塞、感染、炎症等,最终引起支气管扩张和肺实质破坏。如果发生在胰腺和胆道,则造成分泌物干燥并阻塞胆管。如果发生在汗腺,则导致皮肤表面的氯和钠的浓度升高,有的患者出汗时皮肤表面可看到氯化钠结晶。该病通常在婴幼儿时期发病,新生儿出生后数日内即可出现症状,表现为反复支气管感染、气道阻塞、轻度咳嗽,并伴发肺炎、肠黏液分泌和黏度增加、肠梗阻、直肠脱垂等。
随着分子生物学的进展及基因检测水平的不断提高,基因检测技术在囊性纤维化诊断中地位越来越重要。截至2017年,公开报道的CFTR基因突变有2 000多种,但奇怪的是,并不是所有的CFTR基因突变都能引起囊性纤维化,其具体原因尚不明确,相关的研究还在进行中[4]。Li等[5]于2006年报道了我国首例经基因检测验证的囊性纤维化患儿。患儿确诊年龄14岁,自生后即开始出现反复咳痰,7岁时确诊为支气管扩张症,并且合并有鼻窦炎、中耳炎、脂肪泻、皮肤氯化钠结晶等,CFTR基因检测证实其存在分别遗传自父母的复合杂合突变。
本报道中2例患儿系兄妹,均自生后出现反复咳嗽、咳痰,妹妹既往有多次“脱肛”史,自3岁起进食油性食物后出现脂肪泻,合并中心性支气管扩张症、肝硬化、胆汁淤积、胰腺萎缩并脂肪化、血糖高、副鼻窦炎、过敏性支气管肺曲霉病;哥哥自生后反复咳嗽,合并中心性支气管扩张症、脂肪肝、胰腺萎缩并脂肪化、副鼻窦炎、出汗时皮肤表面可见氯化钠结晶。通过基因检测,兄妹二人均确诊囊性纤维化。本文中的两个基因突变类型均已有文献报道[6-8]。但不同的突变类型是否导致不同的临床表型需有待于进一步研究证实。
既往的研究发现,亚裔囊性纤维化的CFTR基因突变类型与西方有很大差异。在我国囊性纤维化患者中出现频率最高的为c.2909G>A/G970D位点,在欧美人中该位点突变却很少见;c.1766+5G>T/1898+5G>T我国已有6例报道,但在欧美囊性纤维化患者中却未见报道[8-10]。因此,对于临床症状怀疑囊性纤维化的患者均应行全外显子测序,以便发现少见的基因突变甚至新的基因突变,丰富囊性纤维化基因谱。
囊性纤维化治疗主要是对症治疗,包括防止感染,清理气道,改善呼吸,并维持足够的营养,防止营养不良。近年来,随着分子生物学的快速发展,使得囊性纤维化的治疗有了重大突破。由于囊性纤维化是单基因疾病,基因治疗可以针对CFTR基因编码的蛋白功能缺陷从根本上治疗囊性纤维化。最初的囊性纤维化基因疗法是采用病毒或非病毒载体,试图将健康人的CFTR基因转染到CFTR基因缺陷的细胞中,但是相关的临床试验尚未显现明显的效果。Ivacaftor于2012年美国食品药品监督管理局批准上市[11],它是一种CFTR调节器增效器,通过延长CFTR通道开放的时间,来加速上皮细胞表面氯化物的转运,降低分泌物表面氯化钠浓度。患者在应用该药2周后病情即可得到改善,效果可维持48周,同时未见严重的不良反应报道。目前常用的药物包括依伐卡托、芦马卡托等。但遗憾的是,对于中国囊性纤维化患者来说,目前并无针对中国人特有基因型的有效靶向药物。
如不进行干预治疗,囊性纤维化患者预后较差,约半数患儿因并发症于10岁前死亡,活至成年者较少,如能早期诊断并治疗,可存活至成年。随着基因检测技术的发展及多学科联合治疗水平的提高,儿童病死率较前已有明显下降,患者中位预期寿命已超过40岁[4]。由于囊性纤维化可严重影响患者的生活质量和寿命,给予家庭和社会造成沉重的负担,对于已生育该类患者的家庭,再生育时可选择进行第三代试管,优生优育,减少出生缺陷。
猜你喜欢
英语世界(2023年6期)2023-06-30
中国生殖健康(2020年2期)2021-01-18
新疆大学学报(自然科学版)(中英文)(2020年2期)2020-07-25
医学新知(2019年4期)2020-01-02
中国临床医学影像杂志(2019年1期)2019-04-25
小学生导刊(2018年13期)2018-06-29
海南医学(2016年8期)2016-06-08
西南国防医药(2015年7期)2015-02-28
现代检验医学杂志(2015年1期)2015-02-06
长江大学学报(自科版)(2014年12期)2014-03-20
