
镅的化学氧化及电化学氧化研究进展
2023-11-01 06:54:08刘子义王东琪
核化学与放射化学 2023年5期
蒋 慧,颜 欣,刘子义,王东琪
大连理工大学 化学学院,辽宁 大连 116024
核能的可持续发展需要解决核废料库存压力和降低其长期的放射性毒性。从最大程度地减少核废料对环境的长期放射性风险的角度来看,若能使用化学的方法将长寿命放射性核素从待玻璃化的废物中分离出来,并通过嬗变技术将这些分离出的超铀元素在先进的核反应堆或嬗变系统中嬗变为稳定核素,将大大简化核废料的管理。其中,超铀元素中的镅(Am)元素常与镧系元素共存于分离后的残液中,而镧系元素的高中子吸收截面会降低Am的裂变效率,因此Am必须在嬗变前与镧系元素分离[1]。
Am在水溶液中通常以三价离子的形式存在,与三价镧系离子的半径接近(如rAm=1.115 Å,rEu=1.087 Å,1 Å=0.1 nm),化学性质(如氧化态、配位数等)非常相似[2],二者的分离一直是核化学领域的难点之一。如何将Am元素从三价镧系元素中分离出来是核废料后处理过程中亟待解决的关键问题之一。根据软硬酸碱理论[3],二者均为“硬”离子,在水溶液中均更易与含有“硬”供体原子(如O原子)的配体配位,因此,常见的萃取剂(通过氧原子配位)难以分离二者。由于Am的5f、6d轨道相较于镧系元素的4f轨道更具弥散性,前者与含N、S等“软”供体原子的配体间的共价相互作用稍强,从而可借助软硬酸碱理论实现二者的萃取分离。早在20世纪80年代,Musikas等[4]就借助叠氮化物实现了Am(Ⅲ)相较于Eu(Ⅲ)的优先萃取,这引发了一系列寻找新型氮配体的工作,大量的双齿、三齿和四齿杂环氮供体配体被系统地研究[5-7]。然而,这种方法通常会存在分离动力学缓慢、配体不稳定的缺点,同时几乎所有这些配体在萃取过程中均对溶液的pH要求较高(难以从酸度大于0.1 mol/L的溶液中提取An(Ⅲ)),这均阻碍了萃取分离在工业中的实际应用[8]。
Am的多价态特性为通过调控Am氧化态来实现镅镧分离提供了可能,作为液-液萃取的替代方法,Am的氧化分离因其高效率和较大的改进空间而受到越来越多的关注。本文在介绍目前对镧锕离子及其配合物的电子结构和氧化还原性质的认识的基础上,简要综述了近年来国内外面向镅锕分离的Am的化学氧化及电化学氧化的研究进展,为建立通过调控价态实现镅镧分离的新分离途径提供思路。
1 Am氧化的结构基础
锕系和镧系元素的特征在于逐步填充的5f和4f电子壳层。其中,锕系元素尤其是前锕系元素,其5f和6d电子具有十分接近的能级,同时锕系元素中的5f轨道相对弥散。这些特点使得锕系元素具有较镧系元素更丰富的价态,如表1[9-16]所示。

表1 已报道的镧系和锕系的氧化态[9-16]Table 1 Accessible oxidation states(OS) of lanthanides and actinides[9-16]
锕系元素的多价态带来了非常复杂的氧化还原行为,如Pu的四个氧化态可以在溶液中平衡共存。Np、Am的最高氧化态可达到+7,Pu的最高氧化态可达到+8,这些高价的物种的稳定性从Np到Am显著降低,因此,尽管在1950—1960年已发现Am在溶液中可被氧化成更高的氧化态(Ⅳ、Ⅴ、Ⅵ价),相比于对Np、Pu等元素氧化还原性质的研究,对处于5f系列元素中间位置的Am元素的氧化-还原化学的研究很少。根据文献,Asprey等[17]在1951年首次使用过硫酸胺将Am(Ⅲ)完全氧化为Am(Ⅵ)。随后,Werner等[18]使用次氯酸钠氧化分离出了高价态的Am(Ⅴ)。此后,由于高价态的Am在水溶液中稳定性差,难以捕捉和表征,尤其是Am(Ⅳ),因此对Am的氧化的研究进展很慢。
在水溶液中Am(Ⅳ)极易自发还原为更稳定的Am(Ⅲ)离子[19-20]。尽管在Am(Ⅴ)和Am(Ⅵ)的还原和歧化反应的研究中推测Am(Ⅳ)作为动力学反应中间体存在,但直到1961年Asprey和Penneman[21]才首次通过将Am(OH)4溶解在15 mol/L氟化铵溶液中得到了稳定的Am(Ⅳ)水溶液,其吸收光谱与固体AmF4相似。作者也发现用饱和的KF、RbF溶液溶解Am(OH)4也可以提高Am(Ⅳ)的稳定性。这些结果表明,在溶液中高浓度的F-可以抑制溶液中还原性物种与Am(Ⅳ)物种的反应,稳定Am的+4价态。
1981年Morss等[22]报道了通过测量AmO2的生成焓等热力学数据计算得到的Am(Ⅳ/Ⅲ)的标准氧化电势数据,即φ(AmⅣ/AmⅢ)=(2.62±0.09) V。1985年Martinot等[23]总结了基于实验获得的不同条件下Am各氧化/还原电对的氧化电势,绘制了Am的氧化电势图(图1)。其中,Am(Ⅳ/Ⅲ)是Am氧化为更高价态时的决速步骤(φ=2.62 V)。

参比电极为饱和甘汞电极(SCE)图1 酸性溶液中Am的标准电极电势[23]Fig.1 Latimer diagram for Am in acid solution[23]


表2 Am(Ⅲ,Ⅳ,Ⅴ,Ⅵ)水合物结构示意图[25]Table 2 Schematic diagram of the structures of hydrated Am(Ⅲ, Ⅳ, Ⅴ, Ⅵ) complexes[25]
相较之下,镧系元素在水溶液中常保持三价(如La、Sm、Gd、Er等),部分可被氧化为四价(如Eu、Ce、Pr、Tb等)[26-29]。这为镅-镧分离提供了一种新的策略,即借助高价的线性镅酰离子(Am(Ⅴ)和Am(Ⅵ))具有与Ln(Ⅲ)/Ln(Ⅳ)截然不同的空间结构和配位化学的性质,利用溶剂萃取或其他分离方法实现它们的有效分离。
2 Am的化学氧化
2.1 过硫酸盐氧化
(1)
过硫酸盐在水溶液中不稳定,在稀硫酸溶液中分解释放出O2,在浓硫酸溶液中形成H2O2。1951年Kolthoff和Miller[30]对过氧二硫酸盐的分解反应动力学进行了研究,提出其在稀硫酸中发生分解反应:
(2)
而在强酸性溶液中:

(3)
(4)
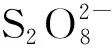
(5)

(6)
其中:Kh,过氧二硫酸氢的解离常数;k1和k2分别代表Ag+的非催化(c(AgNO3)=0)和催化路径的速率常数,对应的活化能分别为28.6、17.4 kcal/mol(1 cal=4.18 J)。

然而,在核废料的储存领域,+2~+6氧化态的硫物种的存在会产生副产物硫酸钠,使得后续玻璃化过程复杂[35]。硫酸盐在硼硅酸盐玻璃中的溶解度低,其溶解度通常只有约为1%SO3(质量分数,含硫组分在该玻璃中的存在形式),即使考虑到玻璃组分的可变性,SO3的溶解极限也只在0.5%~1.4%范围内,从而导致废物玻璃化困难[36]。即使硫酸盐的浓度低于其溶解度极限,硫酸盐也会降低玻璃的耐久性。同时低氧化态的硫会加速腐蚀废物包装材料,这最终会影响地下储存库的性能[37]。
2.2 铋酸钠氧化
铋酸钠(NaBiO3)最初被用于Mn的测定,而Co和Ce的存在会对该方法造成致命的干扰,这是因为它们分别被铋酸钠氧化为Co(Ⅲ)和Ce(Ⅳ)[38]。考虑到Co(Ⅳ/Ⅲ)和Ce(Ⅳ/Ⅲ)的电势分别为1.95 V和1.74 V,均高于Am(Ⅵ/Ⅲ)的电势(1.68 V),因此推测Bi(Ⅴ)可能足以将Am(Ⅲ)氧化为Am(Ⅵ)。1969年,Ford-Smith等[39]通过电位滴定法测量了BiⅤ/BiⅢ氧化还原电对的还原电势约为(2.0±0.2) V(式(7))。
(7)

先前的研究表明,使用NaBiO3可以成功实现水相中Am氧化态的调控,但在后续的双相溶剂萃取过程中,有机溶剂的使用使Am(Ⅴ,Ⅵ)极易被还原,有效接触时间有时仅限于几秒钟。2022年,Wang等[44-45]在NaBiO3氧化的基础上,设计了一种在有机溶剂存在下生成并稳定Am(Ⅴ)的新方案。不同于水溶液中添加氧化剂,当Am/Ln的HNO3溶液与含Bi(Ⅴ)的有机溶液接触时,Ln(Ⅲ)离子和Am(Ⅲ)与二甘醇酰胺配体配位而被萃取到有机相中,有机环境中Am(Ⅲ)被Bi(Ⅴ)氧化至Am(Ⅴ)而后被反萃取至水相中(图3),实验在很宽的酸度范围内(1~10 mol/L HNO3)得到了极高的Am/Ln分离因子(>104)。这为核燃料循环中Ln/Am分离的研究开辟了一条新途径。

(a)——两相系统中Am(Ⅲ)/Am(Ⅴ)及Ln的动态循环,(b)——Am-Eu(Ⅲ)的分离结果,(c)——Am-Cm(Ⅲ)的分离结果图3 NaBiO3氧化Am实现Am-Ln分离[44-45]Fig.3 Am-Ln separation through oxidation of Am by NaBiO3[44-45]
2.3 高碘酸铜(Ⅲ)氧化
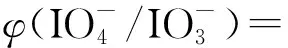
根据McCann等[50]对HNO3溶液中高碘酸铜(Ⅲ)对Am3+的氧化及其在HNO3中的还原动力学的研究,高碘酸铜(Ⅲ)在HNO3溶液中发生自还原,并随HNO3浓度从0.05 mol/L增加到0.5 mol/L而增加,其在酸性条件下的自动还原机制通过解离过程进行。研究发现,在混合前(pH=10.33),高碘酸铜(Ⅲ)主要以[Cu{H2(IO6)2}]5-(或[Cu{IO5(OH)}2]5-)的形式存在,即两个高碘酸盐离子与Cu3+配位,这种配位模式使Cu3+在碱液中能稳定存在。当一个高碘酸根离子从配合物中解离时,还原剂就可以接触到Cu3+并发生配位将Cu3+还原。过量高碘酸钠的加入会改变高碘酸盐阴离子的质子化平衡,导致Cu3+与两个正碘酸盐配体保持配位,从而降低了Cu3+的还原速率。这解释了在Sinkov等[51]的研究中定量氧化的必要性(高碘酸铜(Ⅲ)与Am摩尔比为20∶1)。
在Sinkov等[51]的研究中,在0.25~3 mol/L的HNO3浓度范围内,Cu(Ⅲ)-Am(Ⅲ)恒定的初始摩尔比为10∶1时,98%以上的Am(Ⅲ)被氧化为Am(Ⅵ),但在3.5 mol/L HNO3中,Am(Ⅵ)的转化率只有80%。而增加Cu(Ⅲ)-Am(Ⅲ)初始摩尔比至20∶1,结果98%的Am(Ⅲ)转化至Am(Ⅵ)。相较之下,在3.5 mol/L HNO3中使用NaBiO3氧化Am(Ⅲ),仅有19%转化为Am(Ⅵ)。从结果看,高碘酸铜(Ⅲ)在Am(Ⅲ)的氧化中表现出了比铋酸盐(NaBiO3和Li6KBiO6)更高的反应活性和选择性。
McCann等[50]也研究了镧系离子对高碘酸铜(Ⅲ)氧化效率的影响。他们采用核燃料模拟物为实验体系,发现其中的部分金属离子可与Am(Ⅲ)竞争与Cu(Ⅲ)发生氧化-还原反应,同时,推测由于某一价态的Am被沉淀而仅获得大约81%的Am的质量平衡。这表明了研究氧化分离途径时有必要考虑体系中可能的影响因素。
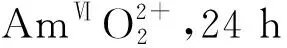
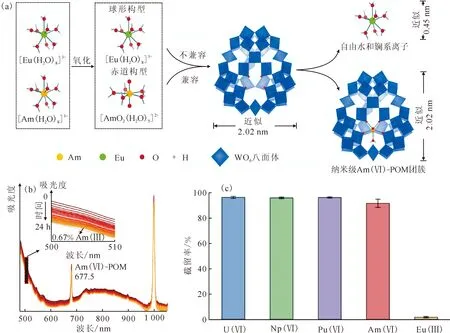
(a)——从镧系元素中超滤分离纳米级Am(Ⅵ)-POM团簇示意图;(b)——在2.0当量的POM存在24 h时,含0.25 mmol/L Am(被高碘酸铜(Ⅲ)氧化)的0.1 mol/L HNO3溶液吸收光谱变化;(c)——U(Ⅵ)、Np(Ⅵ)、Pu(Ⅵ)、Am(Ⅵ)和Eu(Ⅲ)的超滤分离结果图4 通过纳米级多金属氧酸盐簇实现An(Ⅵ)-Ln(Ⅲ)分离[52]Fig.4 An(Ⅵ)-Ln(Ⅲ) separation via nanoscale polymetallic oxygenate clusters[52]
3 镅的电化学氧化
20世纪50年代,Asprey等[17]首次借助铂电极实现了在不同浓度的高氯酸中阳极氧化Am(Ⅲ),在6 mol/L高氯酸中电解1 h后获得最大产率为80%的Am(Ⅵ),但在较低的酸度下,未观察到Am(Ⅵ)。这开创了在不使用任何化学添加剂的情况下,借助电化学相对简单廉价地实现Am(Ⅲ)氧化的先例。Myasoedov等[56]的研究发现,Am(Ⅲ)的电化学氧化的结果依赖于溶液的pH值、阳极电位以及配位介质的性质,其中强配位介质如碳酸盐水溶液有助于稳定高氧化态锕系元素。在3 mol/L KHCO3-K2CO3、pH为8.2~14.0的溶液中电化学氧化Am(Ⅲ),发现Am在阳极电势0.85 V下开始氧化,并在电势达到1.25 V时Am(Ⅲ)的氧化程度达到100%。但当电势进一步增加到1.6 V时,氧化程度下降到0。这与高电势下水的氧化有关,其产物H2O2是高价态Am的有效还原剂(式(8))。
(8)
从反应的动力学看,在3 mol/L K2CO3中的Am(Ⅲ)的氧化有显著的分步氧化的单电子过程特征(图5)。
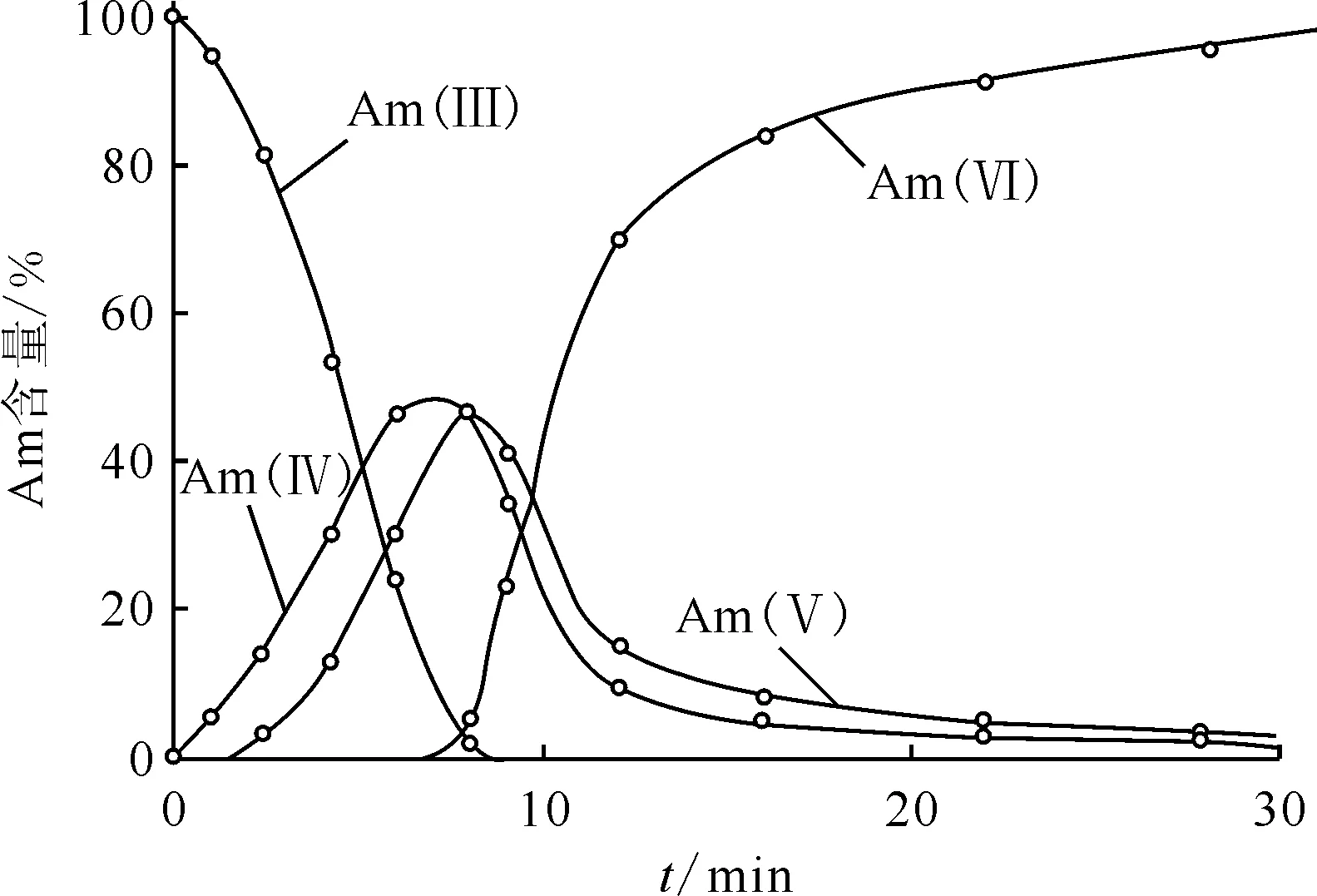
3 mol/L K2CO3,pH=11.3,阳极电势1.15 V,c(Am)=4.23 ×10-4 mol/L图5 Am(Ⅲ)的电化学氧化动力学[56]Fig.5 Electrochemical oxidation kinetics of Am(Ⅲ)[56]


下角a、c分别代表阳极、阴极饱和Na2CO3溶液中的循环伏安图;(b)——计时电流瞬态谱;(c)——阻抗谱[57];裸ITO电极/20 mmol/L HEPES溶液中,在-0.6~0.5 V(实线)、-0.4~0.5 V(虚线)范围内扫描的循环伏安图;(e)——U(Ⅵ)吸附在PM-ITO电极时的循环伏安图;(f)——PM-ITO电极上U(Ⅵ)的阴极峰值电流随ITO电极在磷酸盐溶液中的浸渍时间的变化;(e-f)中的P代表扫描曲线峰位图6 配体修饰电极(AuNPs-CTA及PM-ITO)及裸电极的电化学氧化结果[57,61]Fig.6 Electrochemical oxidation results of modified electrodes and bare metal[57, 61]
对Am的电化学氧化研究的显著进展出现在2015年,Dares等[62]报道了借助4′-磷酰基-(4-苯基)-2,2′:6′,2″-三联吡啶(4′-phosphonyl-(4-phenyl)-2,2′:6′,2″-terpyridine,p-tpy,如图7(a))功能化掺锡氧化铟(tin-doped indium oxide, ITO)电极,在显著更低的氧化电势下(1.8 V vs. SCE)检测到了Am(Ⅴ)和Am(Ⅵ)的生成。实验中使用未官能团化电极在1.8~2.7 V(vs. SCE)下均未观察到Am(Ⅲ)的氧化,而功能化ITO电极在1.8 V的氧化电势下即可观察到一半的Am(Ⅲ)被氧化成Am(Ⅴ)(图7(b))。金属氧化物电极表面结合的p-tpy配体是这种低电位氧化的关键。尽管这一电位下Am(Ⅵ)是热力学可及的,但溶液中还原性中间体的存在导致了Am(Ⅵ)的还原。由于Am(Ⅳ)在溶液中不稳定,在实验中未能研究其配位化学,因此,基于对Pu(Ⅳ)配位化学的理解,以及对Am(Ⅴ)扩散到溶液中之前配合物结构应当保持的推测,作者提出了包含五步的溶液中Am的氧化机理(如图7(c)):(1) Am(Ⅲ)与p-tpy配位;(2) (p-tpy)-Am(Ⅲ)氧化为(p-tpy)-Am(Ⅳ);(3) (p-tpy)-Am(Ⅳ)氧化为(p-tpy)-Am(Ⅴ);(4) (p-tpy)-Am(Ⅴ)水解为[AmⅤO2]+;(5) [AmⅤO2]+氧化为[AmⅥO2]2+。此外,相对于裸-ITO电极,有机配体p-tpy的附着增强了电极表面的疏水性,这最终增加了水氧化过电位,副反应的法拉第效率降低。
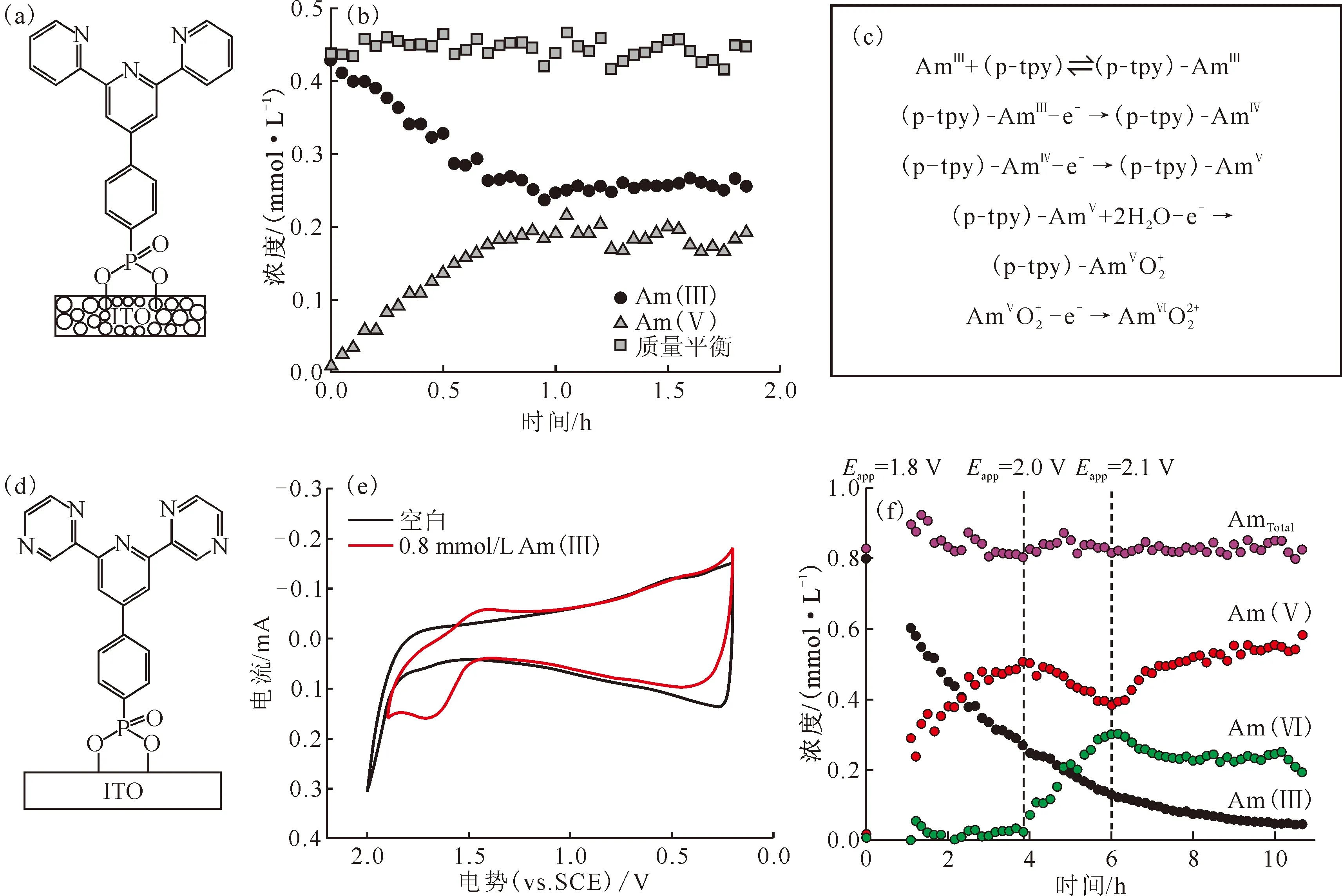
(a)——ITO|p-tpy电极示意图;(b)——Am的种态分布随时间的变化:ITO|p-tpy电极,1.8 V(vs. SCE)施加电势、0.1 mol/L HNO3溶液、0.43 mmol/L Am(Ⅲ);(c)——推测的氧化机理;(d)——ITO|dpp电极示意图;(e)——纳米ITO|dpp电极在0.1 mol/L HNO3中的循环伏安图;(f)——使用纳米ITO|dpp电极电解过程中Am物种浓度变化,Eapp为应用电势图7 Am在配体修饰的ITO电极上的电化学氧化[62-63]Fig.7 Electrochemical oxidation of americium on ligand-modified ITO electrodes[62-63]
在p-tpy功能化电极降低Am(Ⅳ/Ⅲ)氧化电势的基础上,2019年Lopez等[63]进一步将配体拓展到了二吡嗪吡啶(phenylphosphonic acid derivatized dipyrazinylpyridine,dpp,图7(d)),并借助循环伏安法在1.55 V下观察到一个小的Am(Ⅳ/Ⅲ)准可逆氧化还原电对(图7(e)),表明Am的氧化电势被进一步降低。此外,Am氧化产物的分布受施加电势的影响,在1.8 V下,只观察到了Am(Ⅴ),但随着电势增加到2.0 V,Am(Ⅲ)氧化为Am(Ⅴ)随后氧化为Am(Ⅵ),而当施加电势高于2.0 V时,Am(Ⅵ)被还原为Am(Ⅴ)(图7(f)),推测是因为高电势下电极附近水分子发生双电子氧化为H2O2的速率显著增加导致的。这表明在考虑配体修饰电极可降低锕系元素氧化电势的条件下,应同时考虑如何降低副反应如水氧化的效率。
2019年Park等[64-65]将配体修饰电极的策略应用到了镧系元素(Eu、Ce)的电化学氧化领域。
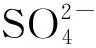
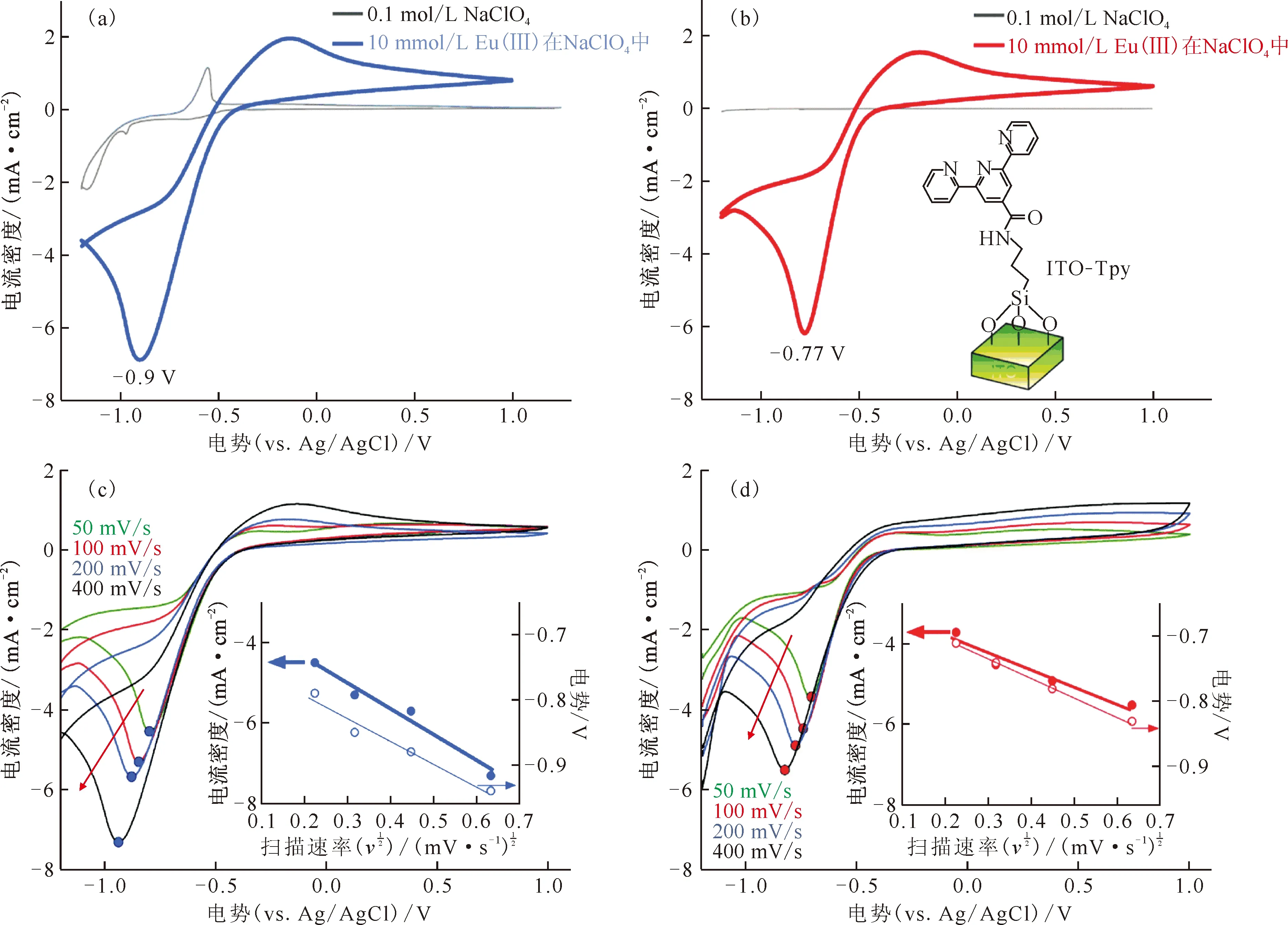
电解液:0.1 mol/L NaClO4和10 mmol/L Eu(Ⅲ)/0.1 mol/L NaClO4(a、b)——200 mV/s扫描速率;(a、c)——裸ITO电极,(b、d)——ITO-Tpy电极图8 Eu(Ⅲ)在配体修饰电极上的电化学氧化[64]Fig.8 Electrochemical oxidation of Eu(Ⅲ) at ligand-modified electrodes[64]
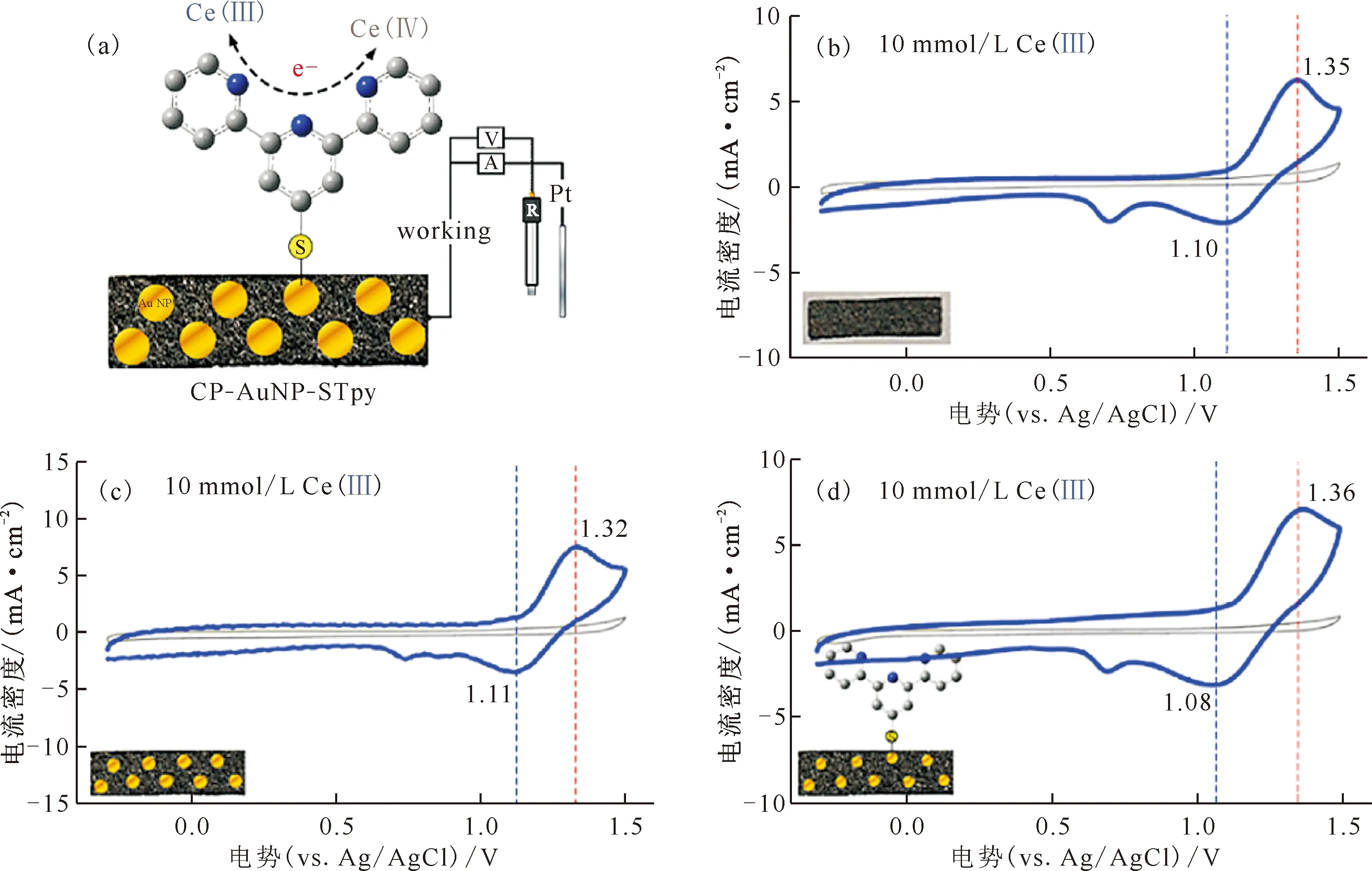
电解质:10 mmol/L Ce(Ⅲ)/0.1 mol/L H2SO4(a)——表面功能化电极CP-AuNP-STpy,(b)——裸CP电极,(c)——CP-AuNP电极,(d)——CP-AuNP-STpy电极图9 Ce(Ⅲ)在配体修饰电极上的电化学氧化[65]Fig.9 Electrochemical oxidation of Ce(Ⅲ) at ligand-modified electrodes[65]
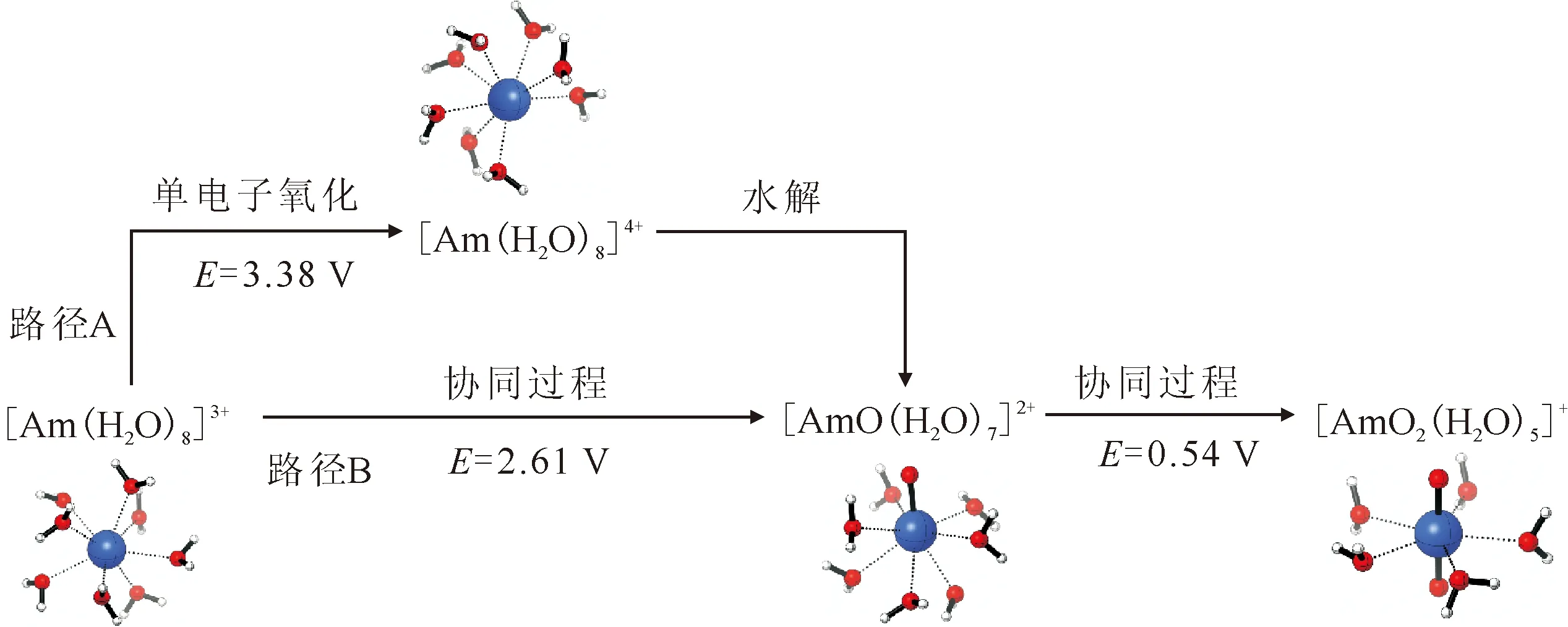
图10 Am在水中的电化学氧化机理[25]Fig.10 Mechanisms of electrochemical oxidation of Am in water[25]
当与含氮配体配位后,由于配体具有比金属离子更低的氧化电位,使反应机理更复杂,进而降低了Am(Ⅲ)到Am(Ⅴ,Ⅵ)的氧化电势。
4 结 论
本文综述了近年来面向镧锕分离的Am的化学和电化学氧化研究进展。研究表明,在Am的化学氧化中,高碘酸铜(Ⅲ)表现出比铋酸盐和过硫酸盐更高的活性和氧化效率,而铋酸盐氧化效率低,过硫酸盐带来的硫物种会影响废液的玻璃化处理,因此高碘酸铜较适合应用于Am的化学氧化。Am的化学氧化结合镧-锕萃取分离技术的研究较为广泛,并获得了极高的分离因子。但有机溶剂的使用易使高价Am还原,开发在有机溶剂存在下生成并稳定高价Am的新方案,或不使用有机溶剂而开发新型的分离方法(选择性配位化合物)是未来化学氧化研究的新方向。近年来有研究致力于将传统氧化剂与其他新型分离技术相结合,有望解决这一问题。
相比于化学氧化面临需要添加强氧化剂、后续处理复杂等问题,Am的电化学氧化提供了可调控的清洁的方案,通过选择合适的配体对电极进行修饰,可显著降低Am(Ⅳ/Ⅲ)的氧化电势,从而避开电解水等影响电化学氧化效率的副反应。由于目前仍缺乏对后续分离效果的研究以及对配体如何参与电化学氧化机制的深刻理解,使得这一方法的工业化应用仍然困难。此外,由于Am的氧化涉及到自还原、歧化等多个过程,需要补充对Am氧化动力学及实验研究中对机理理解的空白,为Am氧化分离策略提供新的见解,助力发展锕系元素氧化分离工艺,同时也有助于发展该策略在其他重金属离子的催化氧化分离中的应用。
猜你喜欢
四川轻化工大学学报(自然科学版)(2021年1期)2021-06-09 06:12:08
防爆电机(2020年4期)2020-12-14 03:11:02
当代陕西(2019年6期)2019-04-17 05:04:10
材料科学与工程学报(2016年4期)2017-01-15 13:35:48
合成化学(2015年4期)2016-01-17 09:01:11
无机化学学报(2014年6期)2014-02-28 17:32:06
无机化学学报(2014年5期)2014-02-28 17:31:42
无机化学学报(2014年4期)2014-02-28 17:31:08
郑州大学学报(理学版)(2013年2期)2013-03-11 20:30:30
新高考·高一物理(2012年5期)2012-04-29 20:27:57
