
超钚元素化合物共价性的理论研究进展
2023-11-01 06:53王聪芝吴王锁石伟群
核化学与放射化学 2023年5期
刘 阳,王聪芝,吴王锁,石伟群
1.中国科学院 高能物理研究所 核能放射化学实验室,北京 100049;2.兰州大学 核科学与技术学院,甘肃 兰州 730000
随着全球能源需求的不断增加,核能作为一种能量密度高、洁净、低碳的能源备受关注。然而核能快速发展所带来的乏燃料后处理问题亟待解决。由于乏燃料中含有极强的放射性和大量未被充分利用的铀和钚,如果采用一次通过循环(乏燃料直接进行地质处置),不仅会浪费有限的核资源,还会对地球环境带来长期威胁。目前,国际上普遍采用的乏燃料后处理方案为PUREX(plutonium uranium reduction extraction)流程[1]联合高放废液(high level liquid waste, HLLW)“分离-嬗变”工艺[2],即将铀和钚等进行分离、回收再利用,之后将废物固化后进行深地质层处置或进行分离嬗变。嬗变处理可将长寿命的次锕系(minor actinides, MA)核素转变为短寿命或稳定的核素,减轻环境负担。
尽管乏燃料中超钚元素Am、Cm的含量很低,但其放射性极强,因此将其从乏燃料中进行分离十分必要。此外,部分超钚元素同位素具有重要应用价值。例如,241Am(半衰期为433 a)是一种优于其它放射性核素的低能量伽马源,主要应用于烟雾探测器和镅-铍中子源(为铍元素(α,n)反应提供α粒子),也是同位素测厚仪和同位素X荧光仪的常用放射源。243Am可用于高中子通量反应堆中生产244Cm、249Bk/Cf、252Cf,以及其他超钚元素[3]。242Cm可以生产更稳定的238Pu,可作同位素电池用于航空、航天等领域[4]。 Bk、Cf同位素是在高通量同位素反应堆中通过一系列中子俘获与β衰变制备的。249Bk可用于制备更重的锕系元素(如能够进行化学研究的249Cf)[5]。252Cf可作为高通量的中子源,用于启动核反应堆、中子活化分析、中子射线照相以及医学治疗等[6]。
尽管目前对铀钍等前锕系元素化合物的研究相对较多,但由于超钚元素制备和观测困难,相关的实验数据十分匮乏,对其结构、化学成键和反应机理的认识非常有限。一般认为,轻锕系元素氧化态比较丰富,随着锕系元素原子序数的增大,5f轨道的收缩使其三价氧化态更稳定,因此超钚元素的性质通常更接近镧系元素[7]。然而最近的报道[8]指出,与已知锕系元素Pu、Am和Cm化合物相比,Cf的硼酸盐具有更显著的共价相互作用,这与镧系元素并不相同。共价相互作用可归因于轨道空间重叠或轨道能量简并,前者在轻锕系元素中更有利。随着锕系原子序数的增大,Bk、Cf化合物5f轨道收缩导致轨道空间重叠有一定程度的降低,但5f轨道能级降低使其与配体轨道发生能级简并的可能性增大[9-13]。
由于研究超钚元素面临高毒性、高放射性且大部分同位素短寿命等挑战,通过实验方法探究相关化合物的结构和性质尤为困难。目前理论计算已成为了解超钚元素物理和化学性质的重要手段。借助于高性能计算平台,通过理论计算和模拟可以揭示超钚元素化合物性质的本质及规律性,进而预测未知化合物的性质,为实验研究提供理论依据[14]。本文结合相关研究结果,对重点探讨超钚元素(主要为Am、Cm、Bk、Cf)化合物共价性的理论研究进行概括介绍,包括无机含氧酸盐化合物以及锕系萃取分离相关配体化合物。
1 超钚无机含氧酸盐化合物

硼酸根是一种极化能力很强的配体,可与锕系元素形成不同结构的硼酸盐化合物[26-27]。美国佛罗里达州立大学的Polinski等[21]先后制备了硼酸镅(Am[B9O13(OH)4]·H2O,AmBO)、硼酸锔(Cm2[B14O20(OH)7(H2O)2Cl],CmBOCl)、硼酸锎(Cf[B6O8(OH)5],CfBO)和硼酸锫(Bk[B6O8(OH)5],BkBO)化合物,其晶体结构如图1和图2[21]所示。在镧系(La-Lu)硼酸盐化合物中Cl-的配位能力越来越弱直至不参与配位,但是锕系元素并不遵循此规律[19]。如果基于纯离子相互作用,性质相似的超钚元素应该形成结构类似或相同的化合物。然而实际上,硼酸锎和硼酸锫的结构相似,但硼酸镅和硼酸锔的结构完全不同。
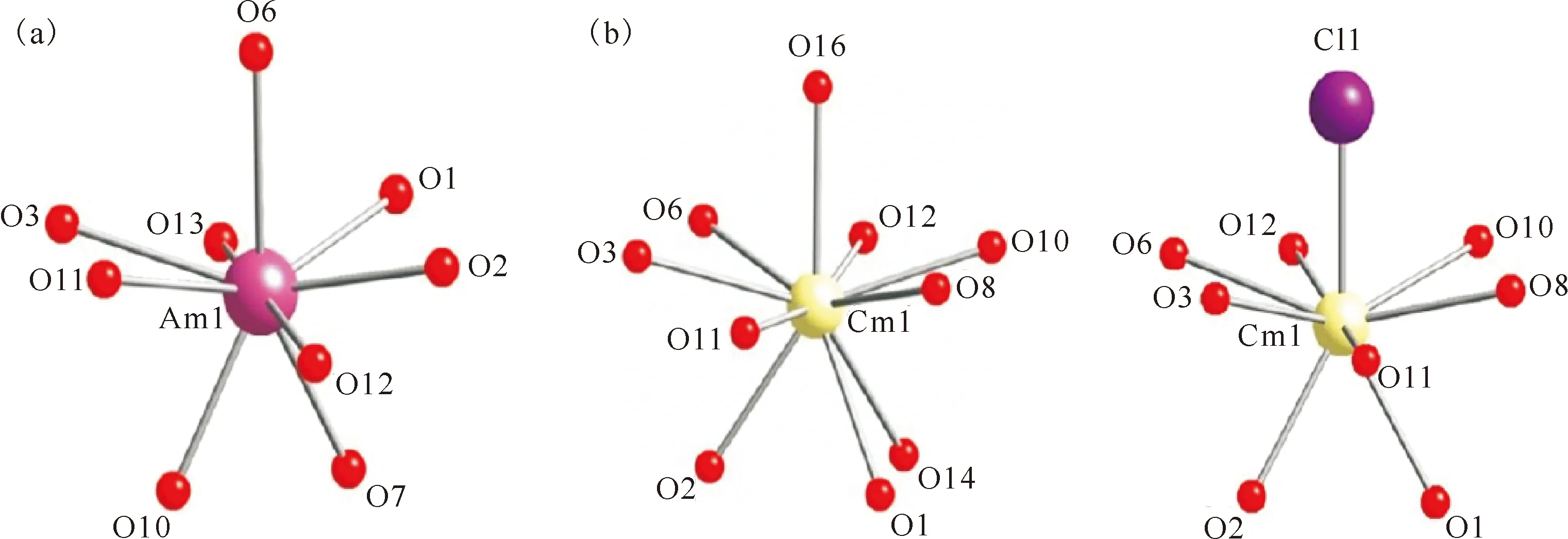
图1 Am[B9O13(OH)4]·H2O(AmBO)(a)和Cm2[B14O20(OH)7(H2O)2Cl](CmBOCl)(b)化合物金属第一配位层的几何结构[21]Fig.1 The geometries of the metal centers of Am[B9O13(OH)4]·H2O(AmBO)(a) and Cm2[B14O20(OH)7(H2O)2Cl](CmBOCl)(b) compounds[21]
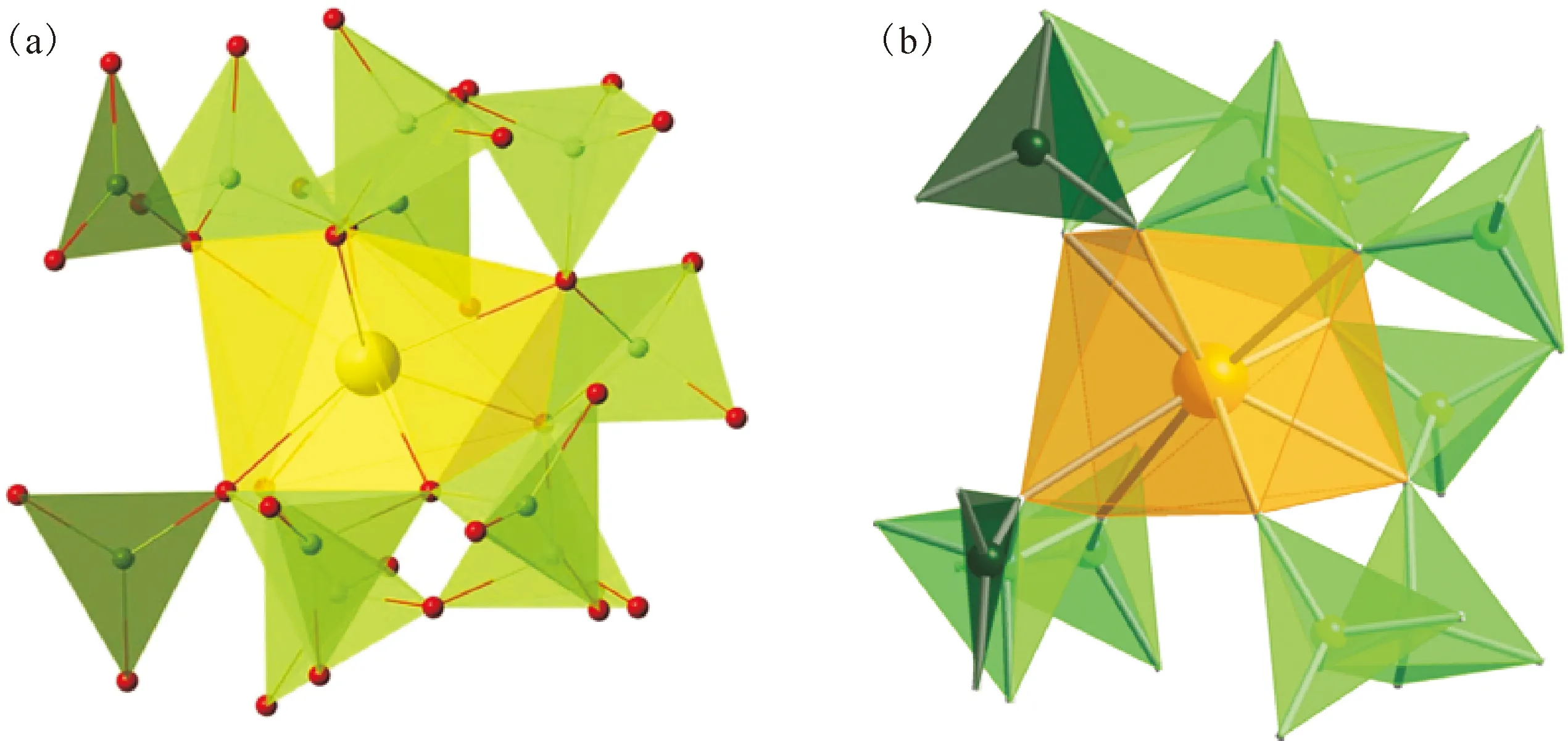
图2 Bk[B6O8(OH)5](BkBO)(a)和Cf[B6O8(OH)5](CfBO)(b)化合物金属中心的几何结构[8,22]Fig.2 The geometries of the metal centers of Bk[B6O8(OH)5](BkBO)(a) and Cf[B6O8(OH)5](CfBO)(b) compounds[8, 22]
密度泛函理论(DFT)研究表明,AmBO的最稳定结构为七重态,而CmBO和CmBOCl为八重态,且Cm为半充满5f7电子构型。自然布居(NPA)的电子构型分析表明,Am和Cm化合物中存在硼酸配位氧原子向锕系离子5f和6d轨道的电子转移。根据完全活性空间自洽场(CASSCF)计算,AmBO和CmBOCl中An的5f轨道均是定域化的,不能与配位原子成键,但6d轨道参与成键,尽管贡献非常小。对CfBO化合物的成键性质分析表明,配体与Cf离子之间存在电子转移,且大部分额外电子位于5f、6d和7p轨道,表明Cf-O键存在共价相互作用特征。CASSCF的计算结果表明,硼酸锎体系中存在一个异常显著的晶体场分裂,至少0.16e从配体转移到5f轨道,这与DFT计算结果一致。因此,这些超钚元素化合物结构的差异与金属5f/6d轨道的贡献有关。
2 超钚元素萃取分离相关配体化合物
锕系元素萃取分离相关配体化合物的配体包括吡啶二甲酸、柔性多齿螯合配体、含硫配体、邻菲啰啉类以及冠醚大环类配体。
2.1 吡啶二甲酸(H2DPA)配体化合物
美国洛斯阿拉莫斯国家实验室Kelley等[9]采用PBE泛函优化An-(DPA)n(An=Am、Cm、Bk、Cf,n=1~3)化合物的结构,同时使用真实溶剂似导体屏蔽模型(COSMO)考虑溶剂化效应,并采用从头算方法对DFT的结果进行验证。H2DPA配体结构示于图3。优化所得1∶3化合物[An(DPA)3]3-的An-O(DPA)键长与An(HDPA)3晶体结构的键长参数吻合较好[22]。An-DPA的键长从Am→Cf逐渐减小,与金属离子的离子半径变化一致。自旋密度分析表明,DPA2-的配位不影响金属中心的氧化态,尽管随着DPA2-的配位,配体的静电斥力增大,An-DPA键长被拉长。1∶1的[An(DPA)(H2O)5]+和1∶2的[An(DPA)2(H2O)2]-化合物为八配位结构,而1∶3化合物[An(DPA)3]3-均为九配位结构。热力学分析表明,Bk和Cf化合物比Am、Cm更稳定,这可能与体系的共价相互作用有关。
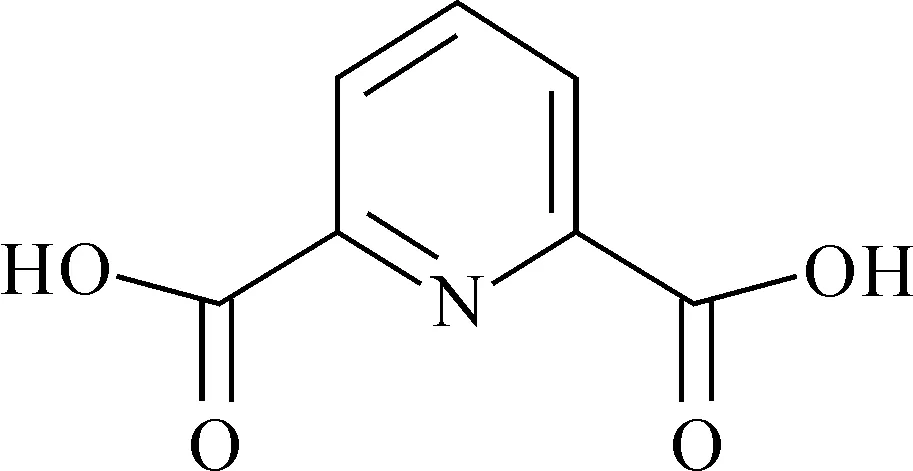
图3 H2DPA配体的结构Fig.3 Structure of H2DPA ligand
图4为[An(DPA)3]3-(An=Am、Cm、Bk、Cf)的分子轨道(MO)图。对于[Am(DPA)3]3-,Am的5f轨道表现出定域化的特征,这些轨道能级大约比DPA配体的高占据轨道高1.5 eV。最高占据分子轨道(HOMO)-最低未占有分子轨道(LUMO)的能隙非常小,只有0.15 eV。从Am→Cf,An的5f轨道的能级逐渐下降,并与DPA配体轨道发生能级简并,导致轨道混合增加,锕系金属的5f轨道更加离域化,从而使金属与配体之间的共价相互作用增强。在Am和Cm化合物中,有6~7个占据轨道具有较多(>80%)的5f轨道贡献。Bk化合物有3个以5f轨道贡献为主的分子轨道,而Cf化合物只有一个类似的分子轨道,但两种配合物中均存在较多5f轨道贡献超过5%的分子轨道。因此,由Am→Cf,5f轨道能级的降低促进其与配体氧原子2p轨道的能级简并。最近,作者报道了[Es(DPA)3]3-化合物[28],实验结果显示,Es化合物的稳定常数高于相应的Cf体系,这说明超钚化合物共价性增强的趋势可以延续到Es体系。
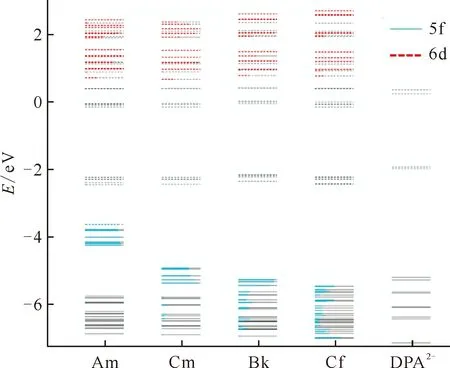
蓝色和红色分别代表An的5f和6d原子轨道贡献,红色或蓝色线的长度与其在分子轨道中的贡献(百分比)成正比;虚线表示空轨道,实线表示占据轨道图4 [An(DPA)3]3-化合物以及配体DPA2-的分子轨道图[9]Fig.4 Molecular orbital diagram of the [An(DPA)3]3- complexes as well as ligand DPA2-[9]
2.2 柔性多齿螯合配体化合物
(1) An-DTPA化合物
二乙烯三胺五乙酸(DTPA)是一种柔性多齿螯合配体(图5),具有三个叔胺氮原子和五个羧酸结合单元,可作为八齿配体参与配位。Deblonde等[29]利用扩展X射线吸收精细结构光谱(EXAFS)结合理论计算首次研究超钚元素化合物An(Ⅲ)-DTPA(An=Am、Cm、Bk、Cf)的溶液化学性质。
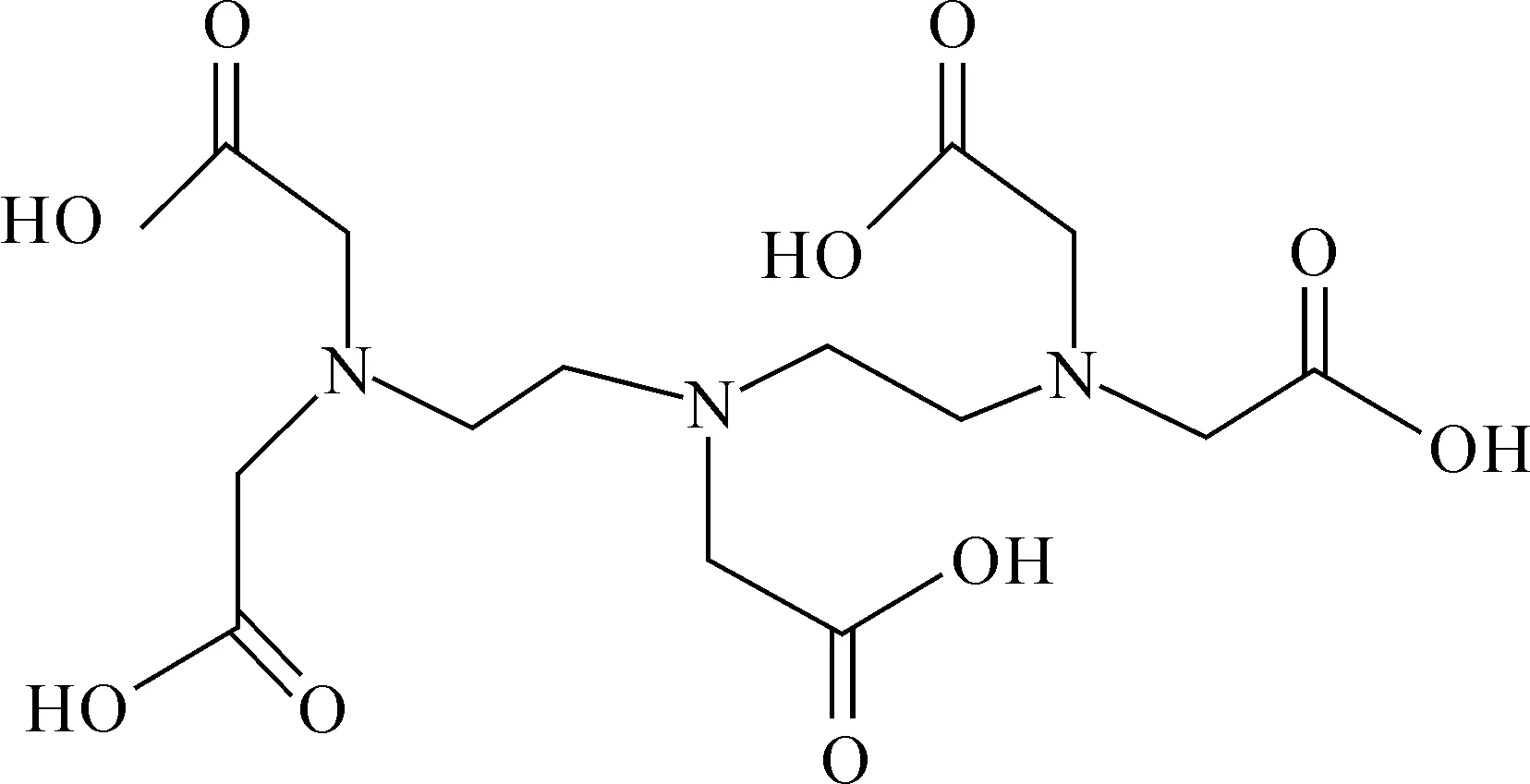
图5 质子化的DTPA配体的结构Fig.5 Structure of protonated DTPA ligand
作者采用PBE泛函优化[An(DTPA)]2-和[An(DTPA)(H2O)]2-化合物的结构。计算结果表明,[An(DTPA)]2-化合物的An-O键长均小于[An(DTPA)(H2O)]2-化合物,且从Am→Cf,An-O键的键长减小。EXAFS分析表明Cf-O键键长比Am、Cm、Bk锕系化合物短。Es(Ⅲ)-DTPA化合物的结构与Cf几乎相同,但Es-O键的键长更短。由于[Cf(DTPA)]2-的Cf-O键距比[Cf(DTPA)(H2O)]2-更接近EXAFS结果,推测Cf、Es化合物较短的An-O键可能是由于配位水分子消失形成无水化合物[An(DTPA)]2-导致的。进一步通过[An(DTPA)]2-+(H2O)9→[An(DTPA)(H2O)]2-+(H2O)8反应进行了验证。计算的Am、Cm和Bk化合物反应ΔG为负值,而Cf和Es化合物的ΔG为正值,表明Am、Cm和Bk 与DTPA配位的产物为[An(DTPA)(H2O)]2-,而Cf和Es以无水[An(DTPA)]2-的形式存在。Cf-DTPA化合物具有较短的Cf-O键,说明Cf体系可能存在更强的共价相互作用,利用这种成键性质的差异DTPA配体也许能够在超钚元素的组内分离中发挥作用。
后续作者研究了超钚元素(Am、Bk、Cf、Es)氨基聚羧酸盐化合物,其中配体为氨基三乙酸(NTA)、N-羟乙基乙二胺三乙酸(HEDTA)、反式1,2环己二氨四乙酸(CDTA)和DTPA[28]。实验结果显示,与Am相比,配体与超钚元素(Bk—Es)的结合常数更大,且配位数越高稳定常数的增加越明显。理论研究以An-DTPA化合物为代表分析锕系氨基聚羧酸化合物的电子结构,采用PBE0杂化泛函优化得到1∶1 An-DTPA化合物的最低能量结构。相对于Am、Bk、Cf,Es的5f轨道与配位O或N原子的2p轨道的能级简并程度更高。Es-DTPA化合物仍遵循配位能力增强的趋势。因此,类似于DPA体系,超钚氨基聚羧酸盐化合物共价性增强的趋势可扩展至Es体系。
(2) An-HOPO化合物
羟基吡啶酮类配体3,4,3-LI(1,2-HOPO)包含四个羟基吡啶酮1,2-HOPO基团,通过多胺骨架连接,与锕系离子能够形成八齿化合物,如图6所示。当完全脱质子时,HOPO带四个负电荷。金属离子可以通过1,2-HOPO单元中的羰基氧原子O(C)和羟基O(N)结合,有报道表明HOPO配体可能诱导金属离子的氧化或还原[30-31]。
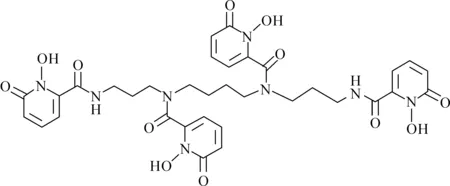
图6 质子化的3,4,3-LI(1,2-HOPO)配体的结构Fig.6 Structure of protonated 3, 4, 3-LI(1, 2-HOPO) ligand
Kelley等[32]采用PBE泛函优化An(Ⅲ)/An(Ⅳ)-3,4,3-LI(1,2-HOPO)(An=Am、Cm、Bk、Cf、Es)化合物的结构,计算的结构参数与EXAFS实验数据吻合较好。对于[An(HOPO)]-,An-O键键长从Am→Cm逐渐增加,Cm→Cf逐渐降低。而[An(HOPO)]0化合物的An-O键键长比[An(HOPO)]-化合物的短。对于配位反应:
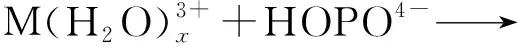
[An(HOPO)]0化合物反应的自由能较[An(HOPO)]-更负,说明An(Ⅳ)与配体HOPO的配位比An(Ⅲ)更有利,这与实验热力学研究结果一致。[An(HOPO)]0中锕系离子在6d轨道上的电子转移与[An(HOPO)]-相当,但在5f轨道上的电子转移明显更高。然而,[An(HOPO)]0化合物在5f轨道上的电子转移增加趋势在Bk处发生逆转,这是由于[An(HOPO)]0化合物中Bk的氧化态为+4价,而Cm和Cf被还原,接近+3价氧化态。四价Bk的5f轨道处于半充满状态,相比于三价Bk更为稳定,所以四价Bk并没有被还原。
图7为[An(HOPO)]-和[An(HOPO)]0化合物的分子轨道分析。由图7可知:尽管在[An(HOPO)]-和[An(HOPO)]0化合物中,An的6d轨道保持在恒定的能级,但An的5f轨道能级降低,与配体轨道的能级逐渐接近,An 5f轨道与HOPO4-配体中1,2-HOPO单元的π轨道简并,导致金属中心与配体之间的轨道混合增强。例如,在[Bk(HOPO)]-中,5f轨道贡献分布在29个分子轨道上,而且每个分子轨道中其贡献在5%以上,即[Bk(HOPO)]0化合物5f轨道与配体轨道的混合逐渐增多。因此,在超钚元素化合物An-HOPO中,随着锕系元素原子序数的增大,化合物变得更加稳定。
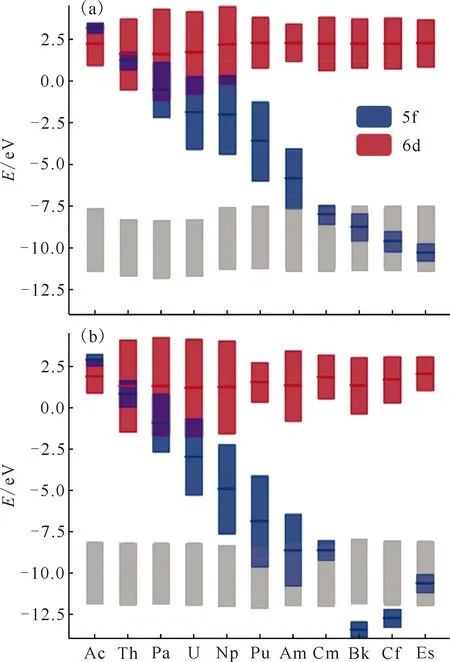
HOPO4-配体(包括吡啶环和配位氧原子)中1,2-HOPO单元的共轭π轨道(灰色);深色的线表示轨道的平均能量值,阴影区域则是标准偏差图7 [An(HOPO)]-(a)和[An(HOPO)]0(b)化合物中An的5f(蓝色)和6d(红色)轨道的平均能级[32]Fig.7 Average energy levels of the 5f(blue) and 6d(red) orbitals in the [An(HOPO)]-(a) and [An(HOPO)]0(b) complexes[32]
(3) An-TPAEN化合物
印度巴巴原子研究中心Ghanty及合作者[33]采用DFT方法研究了水溶性配体N,N,N′,N′-四(6-羧酸-2-吡啶甲基)乙二胺(H4TPAEN)与超钚元素的配合作用。图8为优化的An-TPAEN化合物的几何结构。由图8可知,TPAEN配体通过四个羧酸氧原子和四个氮原子与锕系离子配位。从Am→Cf,An-O和An-N键的键长减小,且键长的减小大于锕系元素离子半径的收缩。能量分解分析(EDA)结果显示,从Am→Cf,静电相互作用降低,而轨道相互作用增强。此外,对配体和金属能级的分析表明,从Am→Cf,锕系元素的能级越来越接近配体的HOMO轨道。该研究结果也说明超钚元素(Bk、Cf)与配体之间共价相互作用增强的趋势。
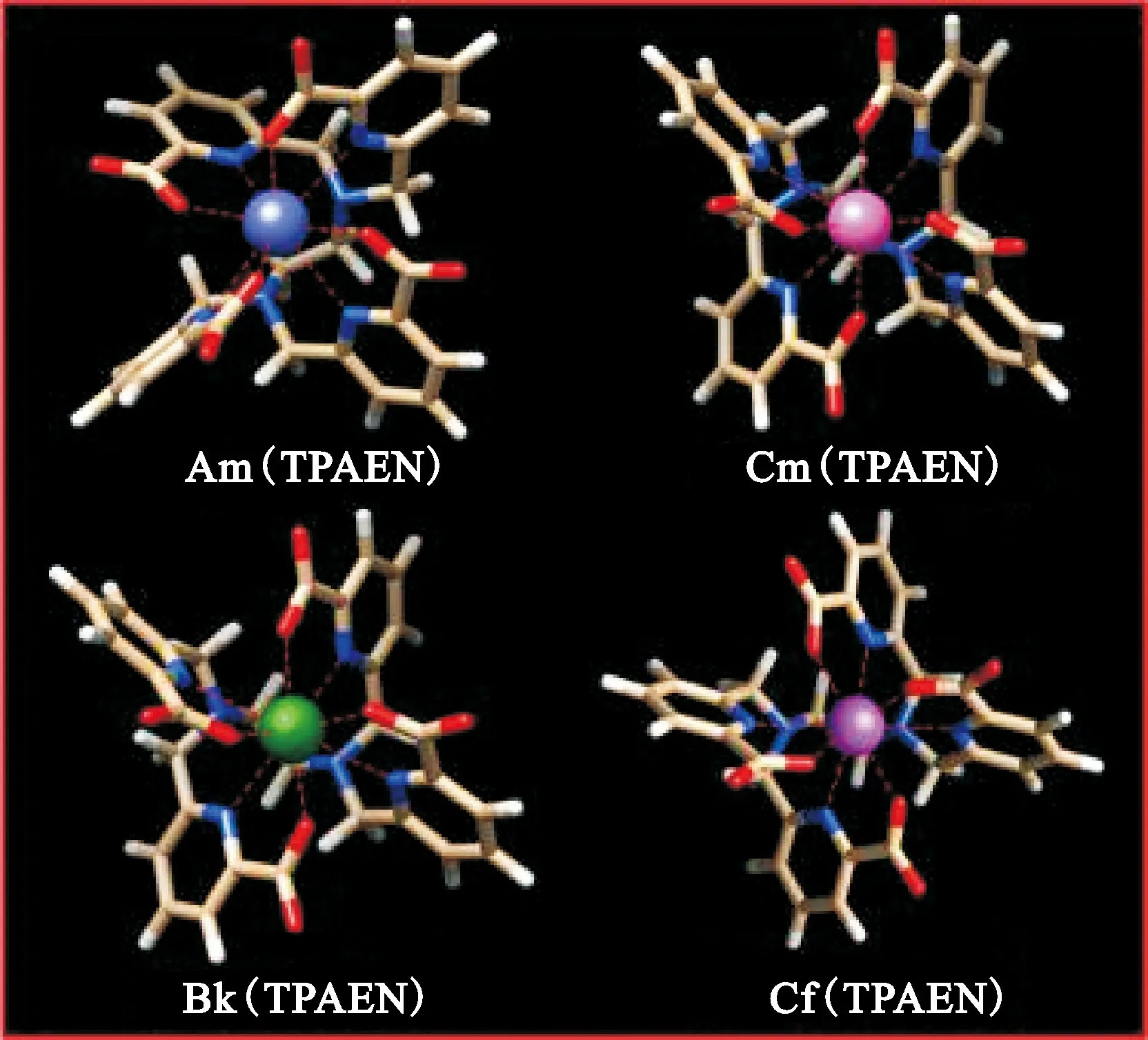
图8 优化得到Am3+、Cm3+、Bk3+和Cf3+与八齿TPAEN配体化合物的几何结构[33]Fig.8 Optimized geometries of Am3+, Cm3+, Bk3+, and Cf3+ complexes with the octa-coordinate TPAEN ligand[33]
2.3 含硫配体An-Cyanex化合物
Ghanty及合作者[33]理论研究了Cyanex类的S、O混合配体与超钚元素Am、Cm、Bk、Cf形成的化合物,并通过EDA方法分析化合物中的共价相互作用。
该工作采用PBE泛函优化了An-Cyanex化合物的结构,计算中使用甲基代替烷基链,对配体进行简化处理,同时采用COSMO隐式溶剂模型在水溶液中对化合物进行模拟。优化得到的中性化合物An(Cyanex)3的结构示于图9。由图9可知:其中Cyanex 301通过两个硫原子与An(Ⅲ)结合,Cyanex 302通过一个硫原子和一个氧原子与An(Ⅲ)结合,Cyanex 272则通过两个氧原子与An(Ⅲ)结合。An-Cyanex化合物中An-O键键长小于An-S键,且键长从Am→Cf呈单调递减趋势,与锕系元素离子半径的减小趋势一致。对An-Cyanex化合物进行EDA分析,结果表明当结合位点由氧转变为硫时,共价相互作用增强,而静电相互作用降低。对于所有的An-Cyanex化合物,从Am→Cf静电相互作用减小,而共价相互作用增强。
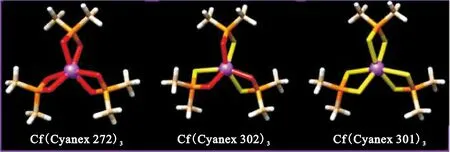
图9 优化得到的Cf与Cyanex 272、Cyanex 302和Cyanex 301配体形成化合物的结构[33]Fig.9 Optimized geometries of Cf complexes with Cyanex 272, Cyanex 302, and Cyanex 301 ligands[33]
2.4 邻菲啰啉类配体化合物
利用DFT方法,对邻菲啰啉类四齿预组织配体N,N′-二乙基-N,N′-二甲苯基-2,9-二酰胺-1,10-邻菲啰啉(Et-Tol-DAPhen)和2,9-二(5,5,8,8-四甲基-5,6,7,8-四氢苯并-1,2,4-三嗪基)-1,10-邻菲啰啉(CyMe4-BTPhen)及其衍生物(邻菲啰啉骨架引入吸电子基团Br和给电子基团phenol)对超钚元素的萃取分离行为进行深入研究[34-35]。采用PBE泛函优化得到AnL(NO3)3和 [AnL2(NO3)]2+(An=Am、Cm、Bk、Cf)化合物的结构(图10和图11)。
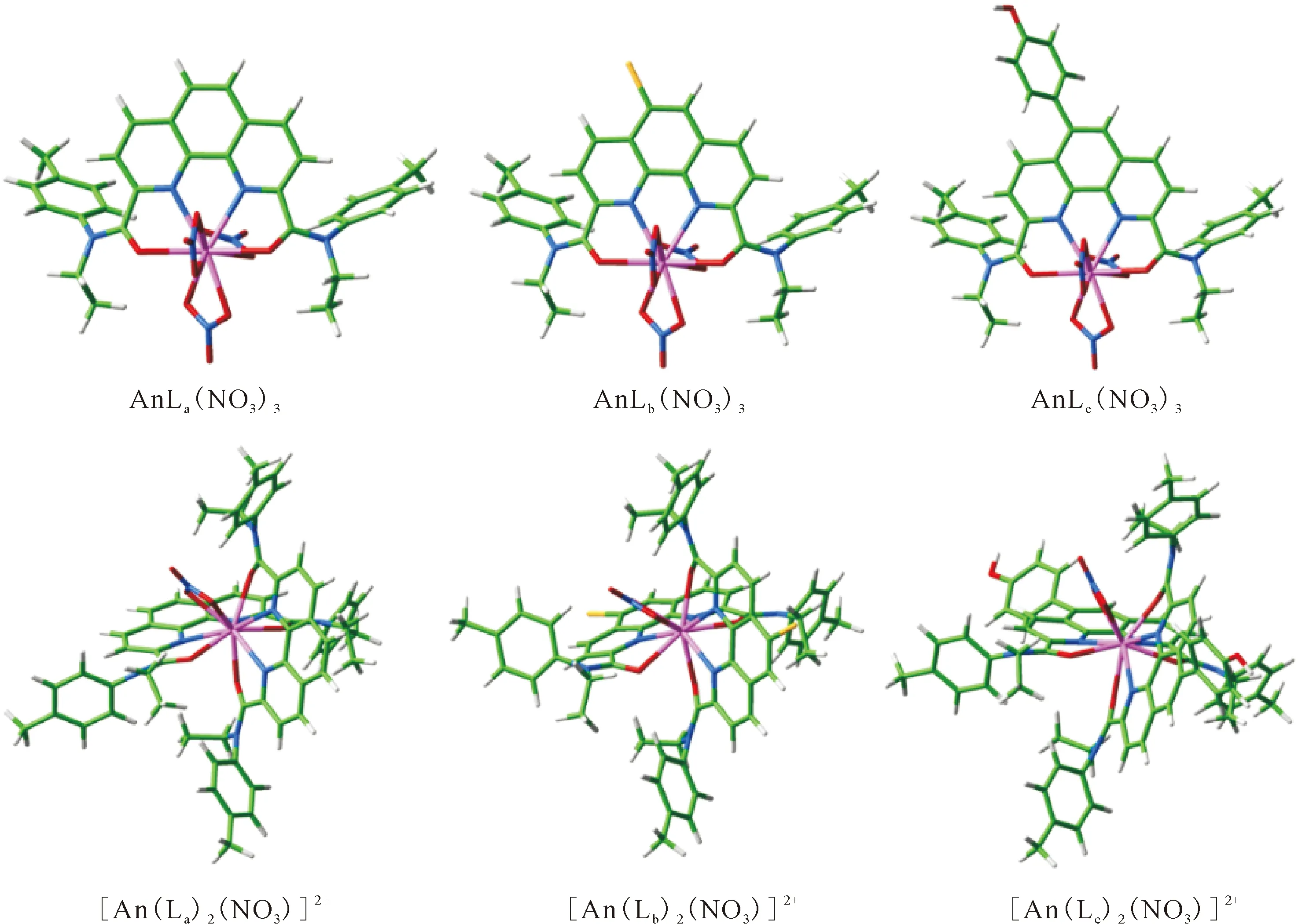
图10 在PBE/6-31G(d,p)/RECP理论水平下优化得到的AnL(NO3)3和[AnL2(NO3)]2+(An=Am、Cm、Bk、Cf)化合物的几何结构[34]Fig.10 The optimized structures of AnL(NO3)3 and [AnL2(NO3)]2+ (An=Am, Cm, Bk, Cf) at the PBE/6-31G(d, p)/RECP level of theory in the gas phase[34]
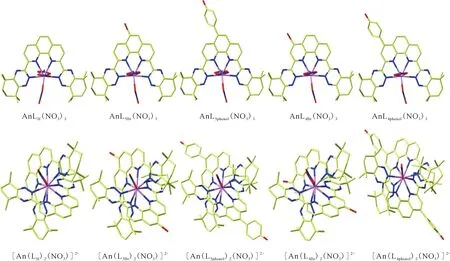
图中H原子省略图11 在PBE/6-31G(d,p)/RECP理论水平下优化得到的AnL(NO3)3和[AnL2(NO3)]2+化合物的几何结构[35]Fig.11 The optimized molecular structures of AnL(NO3)3 and [AnL2(NO3)]2+ at the PBE/6-31G(d,p)/RECP level of theory in the gas phase[35]
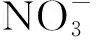
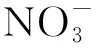
2.5 锕系冠醚大环类化合物
冠醚含有易与金属离子配位的O、N等给电子原子,且不同腔径的大环对不同的金属离子具有配位选择性[36]。该工作[37]采用PBE0杂化泛函优化[Anbp18c6]+和[Anbpp18c6]+(An=Am、Cm、Bk、Cf)化合物结构,系统研究冠醚大环类配体(H2bp18c6和H2bpp18c6)与超钚元素的相互作用以及萃取分离行为。
H2bp18c6和H2bpp18c6配体的结构如图12[37]所示。H2bp18c6配体由4,13-二氮杂-18-冠-6的18元环和两个吡啶-2-羧基组成,而H2bpp18c6配体具有相同的18元环和两个吡啶-2-膦基。在PBE0/6-31G(d, p)理论水平上优化得到具有Ci和C1对称性的配体结构。根据能量比较,发现具有Ci对称性的结构更稳定。以Ci对称性的配体结构为基础,对配体进行性质分析。配体的静电势和分子轨道分析说明H2bp18c6配体对金属离子具有更强的亲和力。化合物的成键分析表明,金属离子与配体配位原子间的共价相互作用从Am→Cf逐渐降低。冠醚大环中的N、O配位原子与An(Ⅲ)的配位能力远弱于侧臂上的吡啶N和羧基/膦基O配位原子,而且侧臂上的吡啶N和羧基/膦基O配位原子与An(Ⅲ)的An-N/O键具有一定的共价相互作用,An的5f和6d轨道主要参与配位。以[An(TMDGA)3]3+为有机相初始物考察该类配体反萃取分离锕系离子的能力,计算结果列入表1。由表1可知:当冠醚配体H2bp18c6的侧链取代基团由羧基变换为磷酸基团(H2bpp18c6)后,配体与锕系离子的相互作用以及配体的萃取能力均有所降低,但配体的组内分离能力增强。由Am→Cf,配体的萃取能力逐渐降低。随着锕系金属离子半径的逐渐减小,冠醚大环骨架对侧臂基团配位的限制作用变强,导致该配体与Cf元素的配位强度变弱。因此,与邻菲啰啉类等配体不同,冠醚配体对超钚元素的配位能力逐渐降低,这主要是由冠醚配体的尺寸选择效应导致的。
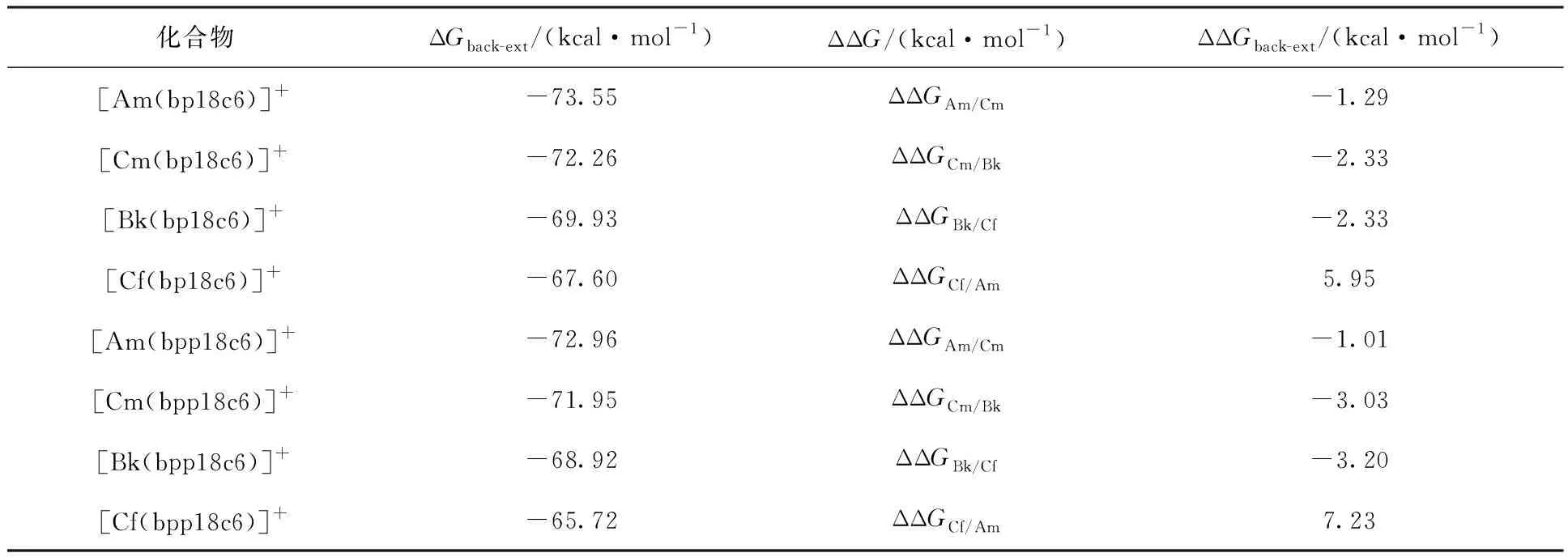
表1 在PBE0/6-311(d,p)/RECP理论水平下计算的[An(bp18c6)]+和[An(bpp18c6)]+化合物反萃反应的ΔGback-ext和ΔΔGback-ext[37]Table 1 The ΔGback-ext and ΔΔGback-ext values for the reactions of [An(bp18c6)]+ and [An(bpp18c6)]+ complexes in aqueous solution at the PBE0/6-311(d, p)/RECP level of theory[37]
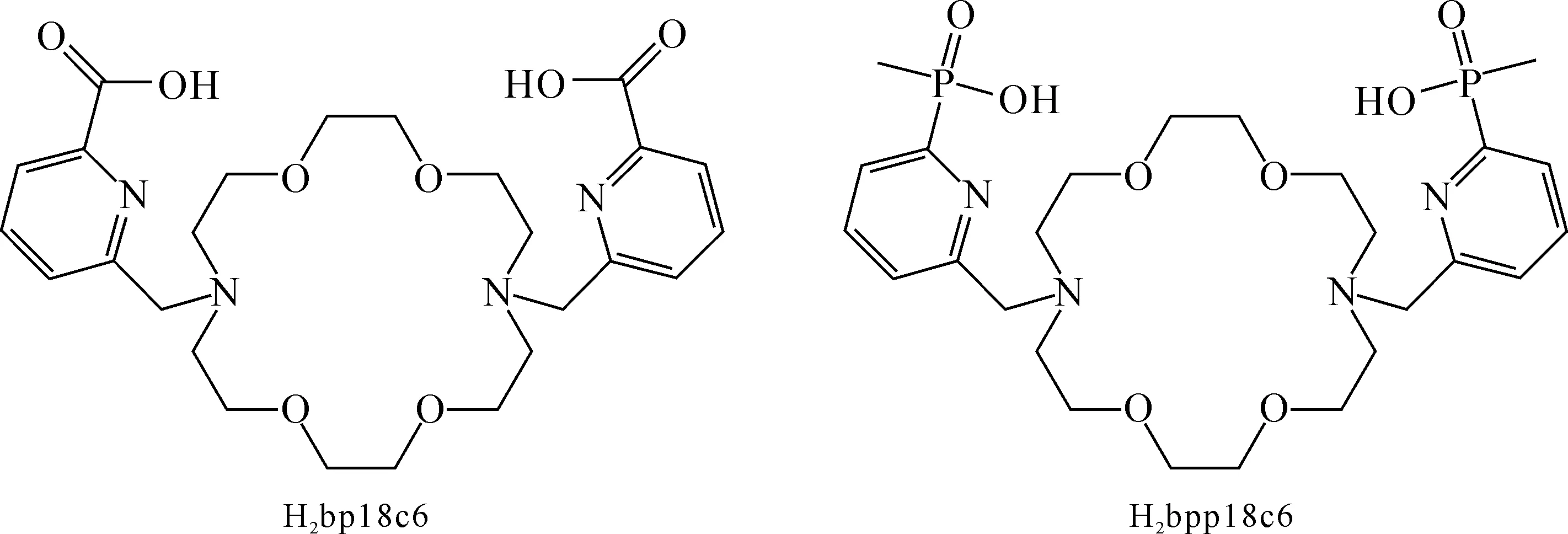
图12 H2bp18c6和H2bpp18c6配体的结构[37]Fig.12 The structures of H2bp18c6 and H2bpp18c6 ligands[37]
3 结论与展望
对近年来报道的超钚元素(Am、Cm、Bk、Cf)化合物的结构和成键性质等理论研究进展进行了综述。实验和理论研究发现,Bk、Cf化合物更强的共价相互作用存在于多种不同的超钚元素化合物中。这些研究结果打破了超钚元素的性质类似于镧系元素这一认知。理论研究表明,超钚元素5f轨道收缩使轨道空间重叠降低,而Bk、Cf化合物5f轨道与配体轨道能级能量简并程度有所增加。尽管如此,由于配位环境的影响,冠醚配体化合物并不遵循共价相互作用从Am→Cf逐渐增强的趋势,这主要是由配体的尺寸选择效应引起的。因此,理论上可以通过不同配体与超钚元素Bk、Cf的配位调节体系中金属与配体的共价相互作用,进而调控配体对超钚离子的萃取和分离能力。由于超钚元素实验研究的局限性,理论研究将发挥关键的作用。目前采用DFT方法对超钚元素化合物的研究表明,PBE和PBE0泛函能够较好地重现实验以及高水平计算方法(耦合簇方法CCSD(T)和基于CASSCF波函数的二阶微扰理论(CASPT2)等)的结果。综上所述,深入了解超钚元素化合物中共价相互作用的差异,探究规律性是今后超钚元素化学研究的重要课题,对超钚元素分离配体的开发具有重要指导意义。
猜你喜欢
杭州(2023年3期)2023-04-03
潍坊学院学报(2021年2期)2021-07-22
高等学校化学学报(2021年7期)2021-07-11
复旦学报(医学版)(2020年3期)2020-06-18
原子与分子物理学报(2020年5期)2020-03-17
青岛大学学报(工程技术版)(2019年2期)2019-09-10
东华大学学报(自然科学版)(2018年1期)2018-06-29
陕西理工大学学报(自然科学版)(2015年4期)2016-01-16
物理化学学报(2015年7期)2015-12-30
中学化学(2015年8期)2015-12-29
