
紫胶皂化废液中回收紫胶桐酸工艺研究
2023-10-30 13:24王世贤唐保山马金菊刘兰香
林产化学与工业 2023年5期
王世贤, 唐保山, 张 弘*, 马金菊, 刘兰香, 徐 涓
(1.中国林业科学研究院 高原林业研究所;国家林业和草原局特色森林资源工程技术研究中心;国家林业和草原局资源昆虫培育与利用重点实验室,云南 昆明 650233;2.南京林业大学 化学工程学院,江苏 南京 210037)
紫胶桐酸是从紫胶树脂中制备得到的一种具有重要价值的天然多羟基脂肪酸,又名罂子桐酸,也称作9,10,16-三羟基棕榈酸,分子式C16H32O5,表观性状为白色晶体[1-2]。紫胶桐酸具有广泛的应用价值,可用于制备防紫外线、耐高温、防辐射的航空航天材料[3-4],也是大环麝香类香料化合物、前列腺素、昆虫信息素、环酰脲、营养能量剂等的重要原料[5-6],具有良好的应用前景。紫胶桐酸通常以皂化、盐析、酸析和重结晶等过程制备[7]。目前国内已有常温皂化法[8]、普通加热皂化法[9]、微波皂化法[10]、超声波辅助皂化法[11]等传统工艺,但传统紫胶桐酸制备工艺仍存在明显的弊端,如反应时间长、条件较苛刻、产率低等[12],并且在制备紫胶桐酸过程中产生大量皂化废液无法处理,只能浓缩后作为危化废品集中填埋。分析表明皂化废液中除大量的环状萜烯外,还有少量的紫胶桐酸。在紫胶皂化后,通过盐析进行紫胶桐酸和其他水解产物的分离,因紫胶桐酸及其钠盐具有一定的水溶性,且水解产物非常黏稠,紫胶桐酸钠盐无法全部析出,使得少量紫胶桐酸钠盐进入滤液而被浪费。在现有的工艺条件下,很难将这部分紫胶桐酸从废液中分离出来。作为天然长链多羟基脂肪酸,紫胶桐酸较为稀缺、珍贵,直接废弃既不经济,也不环保。课题组前期研究表明,紫胶桐酸9、 10位有两个手性中心,存在苏式紫胶桐酸和赤式紫胶桐酸两种构型[13-15],并且两种构型的紫胶桐酸熔点存在较大差异,而且在水相中分散/聚集状态也不同[16]。因此,本研究拟以紫胶皂化废液为原料,通过构型转化法将废液中残留紫胶桐酸转化为赤式紫胶桐酸,再经盐析、酸析、脱色和重结晶进行精制,回收紫胶桐酸,并采用质谱、核磁、TG、FT-IR、DSC、HPLC等方法对其结构和构型进行分析与表征,以期为紫胶加工厂增加效益提供一个可行方案。
1 材料与方法
1.1 原料、试剂与仪器
紫胶皂化废液、苏式紫胶桐酸(纯度>98%),墨江森源科技有限公司;赤式紫胶桐酸,实验室自制[13],纯度98%以上。浓盐酸(质量分数为36.5%)、NaOH、无水乙醇、氯化钠,均为市售分析纯;甲醇、乙腈,色谱纯,加拿大Promptar公司;甲酸,色谱纯,阿拉丁试剂(上海)有限公司。
DSC 200F3差示扫描量热仪、Tensor-27傅里叶变换红外光谱仪,德国Netzsch公司;Agilent 1200型高效液相色谱仪、 1260B型蒸发光散射检测器,美国Agilent公司;超高压液相色谱三重四极杆串联质谱联用仪,英国Waters公司;600MHz核磁共振波谱仪,德国Brucker公司。
1.2 原料成分测定
1.2.1水分和灰分测定 原料中水分和灰分测定参照国标GB/T 8143—2008[17]的方法进行。
1.2.2原料中紫胶桐酸质量分数测定 对紫胶皂化废液进行干燥处理,使用干燥产物配制质量浓度为20 g/L的原料溶液,溶剂为色谱级甲醇,之后过0.22 μm有机系滤膜。然后采用外标法,通过高效液相色谱测量其中紫胶桐酸含量,其中外标法配制质量浓度0.4~0.8 g/L的苏式紫胶桐酸标准品溶液,以峰面积为纵坐标,质量浓度为横坐标进行线性回归分析,线性回归方程为y=5 797.90x-1 362.85,R2为0.999 6,在所测定质量浓度范围内,标准曲线线性良好。色谱条件为:色谱柱Zorbax 5 SB-C18(250 mm×4.6 mm,5 μm),柱温30 ℃,进样量10 μL,流动相采用乙腈-0.1%甲酸溶液梯度洗脱,其中乙腈(5%~100%)60 min,流速1 mL/min。蒸发光散射检测器(ELSD)参数:雾化器和蒸发管温度均为60 ℃,高纯氮气体流速1.6 mL/min。
1.3 紫胶桐酸回收制备
以工厂制备紫胶桐酸的皂化废液为原料,准确称取30 g,加入50 mL的浓盐酸,在100 ℃油浴锅中加热搅拌回流反应7 h使其聚合析出。之后将析出物用大量去离子水超声波清洗以除去游离氯,清洗后干燥备用。取干燥反应物25 g加入300 mL 10%的氢氧化钠溶液,在100 ℃油浴锅中反应约30 h使其完全水解,之后加入过量氯化钠使其饱和进行盐析,抽滤物用饱和食盐水洗涤数次,溶于热水中抽滤除去不溶物,滤液加入20 mL体积分数为10%的硫酸溶液进行酸析,然后抽滤,滤饼在50 ℃下真空干燥制得紫胶桐酸粗品。按照式(1)计算粗品得率(y),并通过外标法计算纯度。
y=m1/m0×100%
(1)
式中:m0—总有机物质量,g;m1—粗品质量,g。
1.4 紫胶桐酸构型转化单因素试验
紫胶桐酸构型转化关键环节在紫胶桐酸氯取代中间体的生成,因此主要研究此步骤操作条件。考察1.3节中原料和浓盐酸料液比(3∶1、 3∶3、 3∶5、 3∶10和3∶15,g∶mL,下同)、反应时间(5、 6、 7、 8和9 h)、反应温度(80、 90和100 ℃)对粗品得率和纯度的影响。
1.5 粗品精制
精制方法采用活性白土进行漂白脱色,20%乙醇溶液重结晶进行纯化。取1 g粗品溶于100 mL 20%乙醇溶液中,同时加入最优用量的活性白土,加热搅拌至粗品完全溶解,继续加热搅拌10 min,然后趁热抽滤除去活性白土,将滤液放置在10 ℃低温下重结晶,28 h后抽滤得到重结晶样品,将样品干燥计算得率并通过外标法计算纯度,多次重结晶以提高纯度。
在1.4节最优方案下大量制备粗品,以活性白土用量为单因素进行脱色,然后以白度为检测指标计算脱色率(η),计算方法见式(2):
η=(d1-d0)/d0×100%
(2)
式中:d0—粗品白度;d1—脱色后样品白度。
1.6 紫胶桐酸的分析与表征
1.6.1红外光谱(FT-IR)分析 采用KBr压片法,压片基质为KBr,压力为10 MPa,波数的扫描范围400~4 000 cm-1,分析谱图中主要特征峰与紫胶桐酸的主要官能团是否一致。
1.6.2质谱(MS)分析 质谱分析采用电喷雾电离(ESI)源,负离子模式,其中毛细管电压为3.5 kV,锥孔电压为70 V,采集模式为4通道选择离子(SIR)采集模式。
1.6.3差示扫描量热(DSC)分析 称取所制备的样品约5 mg进行检测,其中样品坩埚为铝坩埚,保护气氛为高纯氮气,纯度≥99.99%,保护气、吹扫气的流量分别为50和80 mL/min,升温速率10 ℃/min,测量温度范围25~250 ℃。
1.6.4热重(TG)分析 称取制备的样品约5~8 mg进行检测,其中样品坩埚为氧化铝陶瓷坩埚,保护气氛为高纯氮气,纯度≥99.99%,其中保护气和吹扫气的流量分别为50和20 mL/min,升温速率10 mL/min,测试温度范围为室温~800 ℃,降温方式采用自动降温。
1.6.5核磁共振(NMR)分析1H NMR和13C NMR测试溶剂为氘代二甲基亚砜(DMSO-d6),内标为四甲基硅烷(TMS)。
1.6.6C18柱拆分 采用高效液相色谱-蒸发光散射检测(HPLC-ELSD)法,具体条件如下:色谱柱为SB-C18(250 mm×4.6 mm,5 μm),柱温30 ℃,流动相为乙腈和0.1%甲酸溶液,其中V(乙腈)∶V(0.1%甲酸溶液)=25∶75,流速1 mL/min。检测器采用ELSD检测器,其中雾化器温度60 ℃、蒸发室温度60 ℃、气体流速1.6 mL/min。
2 结果与讨论
2.1 原料成分分析
根据1.2节方法测得原料中含水率为27.51%±0.46%、灰分为8.54%±0.84%、游离紫胶桐酸为6.80%±0.06%,通过计算得到原料中含总有机物为63.95%±1.3%。
2.2 不同条件对粗品得率及纯度的影响
2.2.1原料和浓盐酸料液比 浓盐酸和原料中的紫胶桐酸发生聚合反应,产生聚合的紫胶桐酸氯化物,后经氢氧化钠溶液水解产生样品,因此浓盐酸反应阶段对于得率有显著的影响。以30 g紫胶皂化废液为原料,在浓盐酸反应温度100 ℃、反应时间7 h条件下,考察浓盐酸用量对粗品得率及纯度的影响,结果见图1(a)。由图1(a)可知,粗品得率随着浓盐酸用量的增加逐渐增加,纯度则先增后减。当浓盐酸用量小于50 mL时,因为浓盐酸用量少,导致反应进行不完全,因此粗品得率及纯度都较低;当浓盐酸用量大于100 mL时,随着浓盐酸用量增加,使得杂质也有析出,因此粗品得率逐渐增加,纯度逐渐降低。综合考虑得率及纯度,浓盐酸最佳用量为50 mL,因此原料和浓盐酸最佳料液比为3∶5。
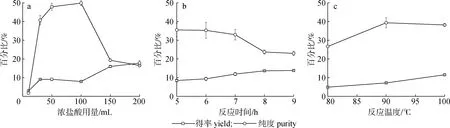
a.浓盐酸用量dosage of HCl; b.反应时间reaction time; c.反应温度reaction temperature
2.2.2反应时间 反应时间是影响得率的一个重要因素,反应时间不足导致反应不完全,从而使得率较低;反应时间过长,样品可能发生其他聚合反应,同时会有杂质析出,因此也会造成样品得率的下降。以30 g紫胶皂化废液为原料,在浓盐酸用量100 mL、反应温度100 ℃条件下,反应时间对粗品得率及纯度的影响结果见图1(b)。由图可知,随着反应时间的延长,粗品得率逐渐增加,纯度逐渐降低。反应时间7 h之前,随着反应时间的延长,得率逐渐增加,纯度下降不明显,说明7 h之前未反应彻底。反应时间7 h之后,随着反应时间的延长,粗品得率增加,纯度降低。综合考虑得率及纯度,选择最佳反应时间为7 h。
2.2.3反应温度 因为原料采用的是工厂废液,反应溶液采用的是浓盐酸溶液,随着浓盐酸的不断反应及挥发,实际上就是水溶液,沸点在100 ℃左右,因此设定反应温度分别为80、 90和100 ℃。以30 g紫胶皂化废液为原料,在浓盐酸用量100 mL、反应时间7 h条件下,反应温度对得率及纯度的影响结果见图1(c)。由图可知,随着反应温度的升高,得率逐渐增加,纯度也呈上升趋势。因此,选择最佳反应温度为100 ℃。
综上所述,确定紫胶皂化废液与浓盐酸的料液比为3∶5、反应温度100 ℃、反应时间7 h,此优化条件下所得聚合产物再经过氢氧化钠水解、盐析、酸洗所得紫胶桐酸粗品的得率为11.53%±0.35%,纯度为38.15%±0.46%。
2.3 粗品精制结果分析
2.3.1活性白土用量的影响 采用活性白土对2.2节优化制备的粗品进行脱色处理,脱色率随活性白土用量的变化趋势如图2所示。从图2可以看出,活性白土脱色效果显著,当活性白土用量(以粗品质量计)为10%时,脱色率达到53.53%。随着活性白土用量的增加,脱色率基本不变,因此活性白土最佳用量为10%。经活性白土脱色1次样品颜色呈现淡黄色,后续脱色应以10%活性白土进行多次脱色,以达到理想的效果。

图2 活性白土用量对脱色率的影响Fig.2 Effect of active clay dosage on decolorization rate
2.3.2纯化得率 紫胶皂化废液经浓盐酸聚合、氢氧化钠水解、盐析和酸析制备紫胶桐酸粗品,粗品经用量为10%活性白土漂白脱色及20%乙醇水溶液重结晶,重复操作1次,即可制备精制紫胶桐酸,制得的紫胶桐酸纯度为80.65%±0.41%,得率为原料质量的3.78%±0.5%,与原料中含有的游离紫胶桐酸(6.80%)相比偏低,原因可能是原料中的游离紫胶桐酸在构型转化制备过程及精制阶段有所损失。在实际生产中,可将精制阶段结晶滤液重复循环利用,这样不仅能提高得率、减少浪费,也能够节约成本、提高收益。
2.4 紫胶桐酸的理化性质
2.4.1FT-IR分析 由红外谱图(图3)可知,纯化产物及紫胶桐酸标准品主要官能团如羟基、羧基及烷基等的振动吸收峰在红外谱图上都有体现,表明其结构的一致性,主要特征峰有3 500~3 000 cm-1处—OH的伸缩振动吸收峰,吸收带强而宽,2 936和2 852 cm-1处为较强的C—H伸缩振动特征峰,1 699 cm-1处为—COOH的振动吸收峰,723 cm-1处为—CH2—的变形振动而且亚甲基数量在4个以上。另外从红外谱图上还可以看出,纯化产物和苏式标准品特征峰存在不同,其中苏式标准品羰基振动吸收峰分裂为1 723和1 661 cm-1,而纯化产物和赤式紫胶桐酸标准品羰基振动吸收峰为单峰,波数在1 699 cm-1处。因此可初步证明纯化产物为紫胶桐酸,且为赤式构型。
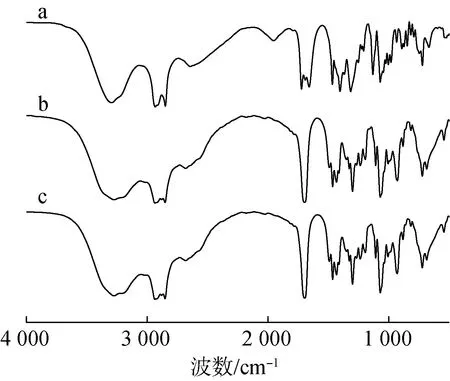
a.苏式thero; b.赤式erythro; c.产物product图3 紫胶桐酸标准品及纯化产物的FT-IRFig.3 FT-IR spectra of aleuritic acid standard and purified product
2.4.2质谱分析 紫胶桐酸是9,10,16-三羟基十六烷酸,相对分子质量为304。纯化产物的质谱分析如图4所示,从图中可以看出,m/z=303处为产物的[M-H]-峰,m/z=339处为产物的[M+Cl]-峰,m/z=607处为产物的[2M-H]-峰,说明产物相对分子质量为304,与紫胶桐酸的相对分子质量一致。由此也可初步判断产物为紫胶桐酸。
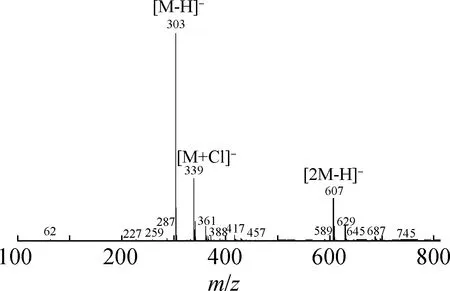
图4 纯化产物的质谱
2.4.3核磁共振分析 纯化产物的核磁分析如图5所示。

图5 纯化产物的13C NMR(a)和1H NMR(b)
从图5(a)可以看出,δ177.56处为—COOH峰,δ73.82和δ73.80处为C-9,10位峰,δ60.78处为C-16位峰,出峰与紫胶桐酸的结构式一致。从图5(b)可看出,产物的氢谱与紫胶桐酸化学式基本吻合。由产物的13C NMR和1H NMR,结合FT-IR及质谱,可判断产物为紫胶桐酸。根据文献[18]及图谱,可将13C NMR和1H NMR图归属如下。
1H NMR(600 MHz,DMSO-d6)δ:11.97(s,1H,OH-1),4.32(s,1H,OH-16),4.19(s,2H,OH-9,10),3.36(t,2H,H-16),3.11(s,2H,H-9,10),2.17(t,J=7.2 Hz,2H,H-2),1.19~1.48(m,22H,H-3,4,5,6,7,8,11,12,13,14,15)。
13C NMR(600 MHz,DMSO-d6)δ:174.56(C-1),73.82、 73.80(C-9,C-10),60.78(C-16),33.71(C-2),32.64(C-8,C-11),32.60,29.28,29.23,28.89,28.62,25.64,25.57,25.46,24.55(C-3,4,5,6,7,12,13,14,15)。
2.4.4TG分析 紫胶桐酸标准品及纯化产物的TG曲线如图6(a)所示,从图中可以看出,苏式紫胶桐酸标准品的最大热失重速率对应的温度为415.8 ℃,产物的最大热失重速率对应的温度为416.9 ℃,两者温度相差较小,说明化学组成的一致性,与之前的FT-IR等表征结果一致,因此可再次印证产物为紫胶桐酸。

图6 紫胶桐酸标准品及纯化产物的TG(a)和DSC(b)
2.4.5DSC分析 纯化产物及标准品的DSC曲线如图6(b)所示,从图中可以看出,产物呈现尖锐的熔融峰,峰光滑且对称,无明显的杂质热吸收峰,其熔融峰值温度为131.89 ℃。有文献报道[14],紫胶桐酸苏式构型的混旋体熔点为99~101 ℃,苏式构型异构单体的熔点为104~105 ℃,赤式构型熔点在124~132 ℃之间。纯化产物与赤式构型紫胶桐酸的熔融热吸收峰呈现高度的一致性,由此可以判断产物为紫胶桐酸,且构型为赤式紫胶桐酸。
2.4.6C18柱拆分分析 采用1.6.6节方法进行高效液相色谱检测,C18柱拆分结果如图7所示。由图可知,纯化产物与苏式紫胶桐酸出峰位置不同,与赤式紫胶桐酸出峰位置相同;同时产物与苏式紫胶桐酸混合时出峰位置保留时间不同,而产物与赤式紫胶桐酸混合时在色谱柱中的保留时间相同,这进一步证明纯化产物为紫胶桐酸,且为赤式紫胶桐酸。
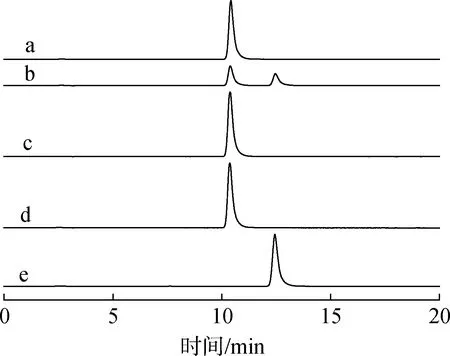
a.产物+赤式product+erytho; b.产物+苏式product+thero; c.产物product; d.赤式erytho; e.苏式thero图7 紫胶桐酸标准品及纯化产物的C18柱拆分高效液相色谱Fig.7 C18 chromatographic HPLC-ELSD spectra of aleuritic acid standard and purified product
3 结 论
以紫胶桐酸制备废液为原料,通过构型转化法将废液中残留的紫胶桐酸转化为赤式紫胶桐酸,再经脱色、重结晶获得赤式紫胶桐酸产品。通过单因素试验优化紫胶桐酸构型转化工艺条件,结果表明:其最佳制备条件为原料和浓盐酸料液比3∶5(g∶mL)、反应温度100 ℃、反应时间7 h,活性白土用量为粗品质量的10%,活性白土脱色及20%乙醇水溶液重结晶各2次可制备精制紫胶桐酸,紫胶桐酸得率为3.78%±0.5%,纯度为80.65%±0.41%。所制备的产物经FT-IR、质谱、核磁、TG等表征证明是紫胶桐酸,并通过DSC、C18柱拆分等表征表明其构型为赤式紫胶桐酸。
猜你喜欢
中国调味品(2022年4期)2022-04-13
云南化工(2021年11期)2022-01-12
昆明冶金高等专科学校学报(2021年1期)2021-05-10
山东冶金(2019年6期)2020-01-06
分析化学(2018年8期)2018-11-01
锻压装备与制造技术(2018年1期)2018-03-28
中国造纸(2015年7期)2015-12-16
中国当代医药(2015年33期)2015-03-01
中国酿造(2014年9期)2014-03-11
大众科技(2013年3期)2013-08-15
