
可见光驱动[FeFe]氢化酶模拟物催化分解水产氢性能研究
2023-10-25 08:22郑会勤樊耀亭
信阳师范学院学报(自然科学版) 2023年4期
郑会勤,樊耀亭
(1. 郑州大学 化学学院, 河南 郑州 450001, 2. 河南财政金融学院 环境学院, 河南 郑州 450046)
0 引言
光催化分解水制氢已成为将太阳能转化为化学能最具潜力的手段[1],其关键步骤是催化剂的选取,因此开发高效光催化产氢催化剂成为研究热点。微生物体内的氢化酶是高效还原质子产生氢气的理想催化剂,这类催化剂是模拟自然界中存在于藻类体内的铁-铁氢化酶的活性位点。2008年,NA等[2]首次报道了一个以[FeFe]氢化酶活性位点 [2Fe2S]模型化合物[(μ-SCH2)2NCH2C6H5] [Fe2(CO)5P(Pyr)3] 为光催化剂、Ru(bpy)32+为光敏剂(PS)、抗坏血酸为质子源和电子给体的三组分光催化产氢体系,该体系在 CH3CN/H2O(V∶V,1∶1) 溶液中,经过3 h的光照,催化转换数TON (turn over number)为4.3。SONG等[3]构建了以[2Fe2S] 模型化合物(5-[(μ-SCH2)2NFe2(CO)6(C6H6)]-10,15,20-三羟基苯基卟啉) 为催化剂、乙硫醇(EtSH)为电子给体、三氟乙酸 (TFA) 为质子源的光催化产氢体系,该体系在CH2Cl2溶液中,以500 W的汞灯照射(λ>290 nm),最大产氢TON只有0.31。ORIGN等[4]报道了一个以[Fe2(μ-bdt)(CO)6](bdt=benzene dithiolate,二硫代苯)为催化剂、曙红Y(EY2-)为光敏剂、三乙胺(TEA)为牺牲剂的光催化产氢体系,该体系在 pH 值为10.5 的 十二烷基硫酸钠(SDS)水溶液中,光照4.5 h的TON为117。WU等[5]报道了多例由氢化酶模型化合物及量子点杂合的光催化产氢体系,并获得了惊人的产氢效率,最高TON可达27 315。
不同于前述列举的活性较高的非均相或杂合产氢体系,本文合成了两个新的[FeFe]氢化酶模拟物1和2,并构建了以化合物1和2为催化剂的三组分均相光催化产氢体系,系统优化了体系的产氢活性,初步探究了可能的电子转移机理,可为开发均相光催化产氢体系提供较高的参考价值。
1 实验部分
1.1 仪器和试剂
Fe(CO)5购自中国科学院兰州化学物理研究所,所用的其他试剂均为分析纯度,化合物1和2均在无水无氧条件下合成。红外光谱采用 Nicolet IR-470型红外光谱仪(KBr压片, 400~4000 cm-1范围内摄谱);元素分析采用意大利FISONS公司的Carlo-Erba1106型元素分析仪;核磁共振采用德国Bruker DRX核磁共振仪(400 MHz);pH值测定采用上海申科pHS-25酸度计;循环伏安采用上海辰华电化学工作站CHI630B测定;荧光淬灭实验采用日本日立HITACHI F-4500荧光光谱仪测定;光催化产氢光源为北京泊菲莱公司的PLS-SXE300/300UV型氙灯;氢气产量测定采用Agilent 4890D气相色谱仪。
1.2 化合物1和2的合成
化合物1和2的合成路线见图1。
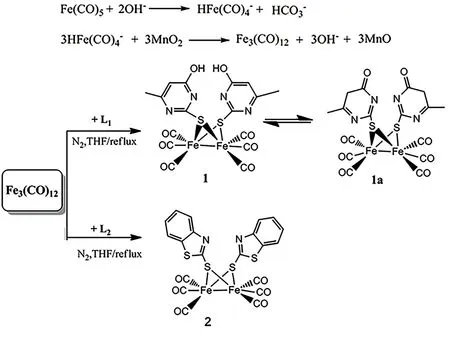
图1 化合物1和2的合成路线Fig. 1 The synthesis routes of complexes 1 and 2
化合物 1的合成: 将0.9 g (1.8 mmol) Fe3(CO)12和0.53 g(3.7 mmol) 4-羟基-2-巯基-6-甲基嘧啶 (L1)置于50 mL的三颈瓶中,向其中加入25 mL干燥的四氢呋喃(THF)作溶剂,氮气保护下将混合物回流搅拌4 h。将反应液冷却至室温并抽滤除去未反应的固体物质,将滤液旋蒸得黑色固体粗产品,该粗产品经硅胶柱分离(CH2Cl2/C6H14=5∶1 作为洗脱剂),收集红色谱带,旋蒸除溶剂并真空干燥后得红色固体产品 0.43 g, 产率为42.5%。元素分析C16H10N4O8Fe2S2:理论值为 C 34.19%、H 1.79%、N 9.97%,实测值为 C 34.56%、H 1.92%、N 1.17%;1HNMR (CDCl3): 2.854(4H,—CH2—), 2.007(6H, —CH3); IR(CO):2070、2039、1999 cm-1,(—C=O) 1645 cm-1。
化合物2的合成: 将0.9 g Fe3(CO)12(1.8 mmol) 和0.64 g(3.8 mmol )2-巯基苯并噻唑 (L2) 置于三口烧瓶中,加入25 mL干燥的THF,氮气保护下将混合液回流搅拌 3 h。后处理方法与1同,得黑红色纯品0.73 g,产率65.8%。元素分析C20H10N4O6Fe2S4: 理论值为C 39.24%、 H 1.32%、N 4.58%,实测值为: C 40.53%、H 1.44%、N 4.75%;1HNMR (CDCl3):δ7.7~7.75(4H), 7.4(4H);IR(CO): 2069、2030、1995 cm-1。
1.3 光催化产氢
光催化反应在一个60 mL的石英反应器中进行。反应时固定反应液为20 mL, 以化合物1、2为催化剂,EBS2-为光敏剂(PS),TEA为牺牲剂和电子给体。光照前,将反应液超声30 min以使溶液均匀,氮气保护并在磁力搅拌下进行光催化反应,反应过程中通冷凝水维持体系的温度为(25±1) ℃,反应液经300 W Xe灯照射(420 nm滤光片滤去紫外光),气相色谱检测生成的氢气浓度 (GC,Agilent 4890D),排水法检测生成的气体体积。
2 结果与讨论
2.1 光催化产氢参数优化
以氧杂蒽醌类有机染料藻红B钠盐(EBS2-)为光敏剂,目标配合物为光催化剂,优化了CH3CN/H2O 的比例、TEA 浓度、溶液的pH及催化剂结构对体系产氢活性的影响,结果如图2。
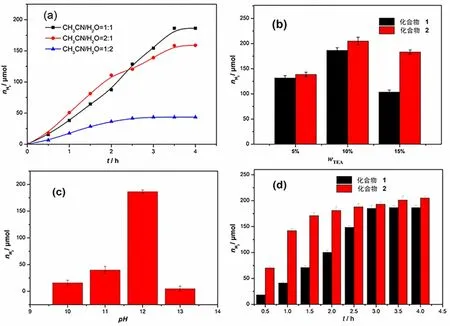
图2 光催化产氢参数优化 Fig. 2 Optimization of photocatalytic hydrogen production parameters
2.2 CH3CN/H2O 的比例对体系产氢活性的影响
探究了几种不同比例的CH3CN/H2O溶液对产氢活性的影响,产氢条件:化合物1 (2.0×10-4mol/L) 、EBS2-(4.0×10-4mol/L),结果见图2(a)。如图2(a)所示,随着CH3CN/H2O (V∶V) 的比例由1∶2变化到 2∶1,体系的产氢活性逐渐降低, 最大产氢量出现在CH3CN/H2O (V∶V)的比例为1∶1 的体系中,经4 h光照,最大产氢量为186.3 μmol (相对于化合物1的TON 为46.6); 处于第二位的是CH3CN/H2O (V∶V) 比例为2∶1 的体系,该体系经4 h光照,最大产氢量为158.9 μmol (相对于化合物1的TON 为39.7);当CH3CN/H2O (V∶V) 比例为1∶2时体系的产氢活性最差 ,经过4 h光照,只有 43.5 μmol (相对于化合物1的TON 为10.9) 的氢气产生。上述结果与文献报道一致[8], 表明溶剂的比例对三组分光催化产氢体系的活性有重要影响,这种现象主要是因为溶剂的性质如极性、介电常数和扩散系数对光催化反应有重要的影响[6]。
2.3 TEA 浓度对产氢活性的影响
进一步探究了TEA 的浓度对产氢活性的影响,产氢条件:化合物1 的浓度为2.0×10-4mol/L, EBS2-的浓度为4.0×10-4mol/L,CH3CN/H2O 的比例为1∶1(V∶V),其结果列于图2(b)。 如图2(b) 所示,当TEA的浓度由5%增加到10%,体系的产氢活性逐渐增大,当浓度为10%时,体系的产氢活性最大,经过4 h的光照,最大氢产量达(186.5±3) μmol(相对于化合物1的TON 为23.3±0.4);当TEA的浓度高于或低于10%时,体系的产氢活性均降低,如当TEA浓度为5%时,体系经过4 h照射,最大产氢量只有(131.6±4.8) μmol(相对于化合物1的TON为16.5±0.6)。这种现象可以解释为:在可见光照射下,EBS2-被激发形成单重激发态的1*EBS2-(经过系间窜越,迅速形成3*EBS2-)和光生空穴,光生空穴迅速将TEA氧化为TEA阳离子自由基[7],这种快速的还原过程使得三重激发态的3*EBS2-能迅速地接受从TEA阳离子自由基转移过来的第二个电子,这一现象与文献报道的一致[8]。
2.4 pH 值对体系产氢活性的影响
探究了溶液的pH值从10升高到13时(用1 mol/L的NaOH和HCl溶液调节溶液的pH值),体系的光催化产氢活性变化情况,产氢条件: 化合物1 的浓度为2.0×10-4mol/L, EBS2-的浓度为4.0×10-4mol/L, TEA的浓度为10%(V∶V),CH3CN/H2O的比例为1∶1(V∶V),结果列于图2(c)。由图2(c)可知,在所考察的pH值范围内,当pH值为12时,体系的产氢活性最高,光照4 h,最大产氢量为(186.5±3) μmol;当pH值大于或小于12时,体系的产氢活性都将显著降低,例如当pH值为13时,体系几乎失去产氢活性,经过4 h的光照,只有痕量的氢气产生。这种现象可能是因为在pH值较低的溶液里(如pH值为10、11) TEA更容易质子化,从而失去部分给电子能力;相反,在pH值较低 (如pH值为13) 的溶液里,不利于产氢活性中间体HFeIIFeI的形成。结果说明,调节溶液的pH值在适当范围内,对目标催化剂光催化产氢活性的提高具有良好的效果。
2.5 催化剂结构对体系产氢活性的影响
为了考察催化剂结构对体系产氢活性的影响,分别以化合物1和2为光催化剂,EBS2-为光敏剂,以10%的TEA为牺牲剂进行产氢实验,其结果列于图2(d)。如图2(d)所示,目标化合物1和2在产氢条件相同的情况下表现出相似的增长趋势,但产氢活性有所不同。以化合物2为催化剂的产氢体系,经4 h光照,其最大产氢量为(205.0±3.5) μmol (相对于化合物2的TON 为 51.5±0.9 );以化合物1作催化剂的体系,最大产氢量为(186.5±3) μmol (相对于化合物1的TON 为41.4±0.4)。结果表明,化合物2的产氢活性高于化合物1。这种结果可能是因为化合物1和2的配体L1和L2所含的质子捕获位点不同,其中L1含4个N原子,L2中含有2个N原子和2个S原子,虽然均含4个质子捕获位点,但L1中含有2个吸电子的—OH,降低了N原子上的电子密度,使其给电子能力减弱,即质子捕获能力减弱,进一步形成氢化物中间体的能力降低。在光催化产氢过程中,[2Fe2S]配合物会形成重要的产氢活性中间体氢化物物种(η2-H2- FeIIFeI),质子捕获能力越强,则越易形成该氢化物中间体[9]。本文报道的两个化合物的产氢活性高于一些文献报道的类似的均相体系的产氢活性[10-11],对构建其他均相三组分光催化产氢体系具有重要的指导意义。
3 可能的电子转移机理
为了探明当前的体系可能的电子转移机理,测试了EBS2-的荧光光谱、牺牲剂TEA对EBS2-的还原淬灭、化合物1和2对EBS2-的氧化淬灭及化合物1和2的循环伏安曲线 。
3.1 荧光淬灭
荧光及荧光淬灭实验结果列于图3(a—d),其中图3(a)为EBS2-的激发和荧光发射光谱,图3(b)为TEA对EBS2-的还原淬灭,图3(c)和3(d)分别为化合物1和2 对EBS2-的荧光淬灭。

图3 EBS2-的激发和荧光发射光谱及荧光淬灭光谱图 Fig. 3 Excitiation, emission spectra and fluoresence quenching spectra of EBS2-
如图3(a)所示,在554 nm处EBS2-出现了较强的荧光吸收峰,说明所选的光敏剂对可见光有很好的吸收。由图3(b)可知,当将TEA加入EBS2-的溶液中时,单重激发态的光敏剂(1*EBS2-) 的荧光发射强度基本保持不变,如当加入3000倍当量的TEA时,EBS2-的荧光发射峰的强度仅有很微弱的降低,表明从TEA到1*EBS2-的直接电子转移是不可行的。这一结果间接证实了该体系的还原淬火只能经由1*EBS2-经系间窜越 (ISC) 产生的3*EBS2-物种进行,这与早期的报道一致[12]。然而,当将不同浓度的目标化合物1和2 (浓度分别从0 mol/L增加到3.0×10-4mol/L和6.0×10-5mol/L) 分别加入EBS2-的溶液中,EBS2-的荧光强度都表现出不同程度的淬灭,当向EBS2-的溶液中分别加入15倍和3倍当量的催化剂1、2时,其荧光强度分别被淬灭了约60.0%(图3(c)) 和88.9%(图3(d))。两个催化剂的淬灭常数 (kq) 可分别根据Stern-Volmer 方程[13]计算(方程(1))。
I0/I=1+kqτ0[Q],
(1)
式中:I0是不加淬灭剂时EBS2-的荧光强度,I为加了淬灭剂时的荧光强度,τ0为无淬灭剂时EBS2-的荧光寿命 (τ0=75 ps[14]),kq为淬灭速率常数,[Q]为淬灭剂的浓度。由式(1),可计算出目标化合物1和2对EBS2-的荧光淬灭速率常数kq1和kq2分别为6.7×1013、1.8×1015L/(mol·s),这两个值均大于2.0 × 1010L/(mol·s),说明该荧光淬灭是双分子的静态淬灭;同时,较大的荧光淬灭常数表明化合物1和2对EBS2-的荧光均产生快速的淬灭,即从1*EBS2-到化合物1和2的第一个电子的转移均能快速地进行。
3.2 化合物1和2的电化学性能
在乙腈中分别测试了化合物EBS2-、1和2的电化学行为,循环伏安曲线(CV)和相应的电化学数据分别列于图4(a—c)和表1。由图4(b)和4(c)可知,两个样品的CV曲线都出现两个不可逆的还原峰,分别为-0.80、-1.91 V(1)和-1.11、-1.46 V (2), 这两个还原峰分别归属于从 FeIFeI到FeIFe0和FeIFe0到Fe0Fe0的单电子还原过程;同时,两个化合物也分别在+2.35 V和+2.26 V处出现了一个不可逆的氧化峰 (表1),对应于 FeIFeI到 FeIFeII的氧化过程。

表1 EBS2-和化合物 1、2 在CH3CN溶液中的还原电位及热力学数据Tab. 1 Reduction potentials and thermodynamic data of EBS2-, complexes 1 and 2 in CH3CN solution
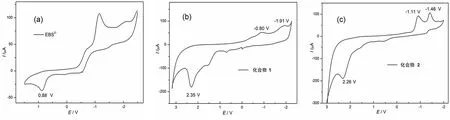
图4 光敏剂EBS2-(a)、化合物 1(b) 和2(c) 的循环伏安曲线Fig. 4 CV grams of EBS2-(a), complexes 1 (b) and 2(c)
值得注意的是,化合物1和2的第一还原峰的电位比CHONG等[15]报道的(μ-pdt)[Fe(CO)3]2更正,表明它们更容易从光敏剂接受电子并将电子传递给质子产生氢气。由EBS2-的氧化电位(Eox)、化合物1和2的还原电位(Ered)及EBS2-激发态的能量E00(E00=hc/λ00,λ00=542 nm,则E00=2.29 eV),根据Rehm-Weller 式(2)[13],可求出电子从光敏剂转移到催化剂时的自由能(GΘ)变化。
ΔGΘ=Eox-Ered-E00。
(2)
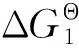
4 结论
合成并表征了两个新的 [FeFe]氢化酶模拟物1和2,构建了以目标化合物1和2为光催化剂,EBS2-为光敏剂,TEA为电子给体和质子源的三组分光催化产氢体系,该体系在pH值为12 的体积比为1∶1的CH3CN/H2O溶液中,经可见光照射(λ>420 nm)4 h,最大产氢量分别为(186.5±3)μmol(相对于化合物1的TON 为 41.4±0.4)和(205.0±3.5) μmol(相对于化合物2的TON 为51.5±0.9)。结果表明,配体中含有较多的质子捕获位点,有利于形成产氢活性中间体H2-2Fe2S(η2-H2-FeIIFeI)物种,从而提高催化剂的产氢活性。荧光淬灭实验和电化学研究表明,光生电子从1*EBS2-到化合物 1和2的第一个电子的转移均为热力学可行过程,到化合物 1和2的第二个电子转移是热力学不可行过程。化合物1和2将是潜在的光催化产氢催化剂。
猜你喜欢
化工学报(2021年1期)2021-01-30
山东化工(2019年2期)2019-02-16
陶瓷学报(2019年5期)2019-01-12
福建农林大学学报(自然科学版)(2018年5期)2018-10-11
无机盐工业(2017年5期)2017-05-25
郑州大学学报(理学版)(2017年1期)2017-04-07
三峡大学学报(自然科学版)(2017年1期)2017-03-20
化工管理(2017年25期)2017-03-05
中国资源综合利用(2016年9期)2016-01-22
应用化工(2014年7期)2014-08-09
