
儿童肾透明细胞肉瘤3例临床病理分析并文献复习
2023-10-16 07:26:52张昕婷陈余朋班超然
临床与实验病理学杂志 2023年9期
张昕婷,陈余朋,班超然,张 声,陈 虹
儿童肾透明细胞肉瘤(clear cell sarcoma of kidney, CCSK)是常见的儿童肾脏恶性肿瘤,占儿童肾肿瘤的4%~5%[1],具有骨转移及脑转移倾向,也可扩散至肺部及腹盆腔,其侵袭性的生物学行为及晚期复发倾向是预后不良及高病死率的主要原因[2]。CCSK的组织学形态多样,缺乏特异性免疫组化标志物,诊断困难,本文回顾性分析3例CCSK的临床病理学特征、免疫表型、分子遗传学特点,复习相关文献,旨在提高临床和病理医师对其的认识水平,减少误诊、漏诊。
1 材料与方法
1.1 材料收集2015年1月~2018年12月福建医科大学附属第一医院诊断的3例CCSK石蜡标本,由高年资病理医师重新阅片确诊,并对患者进行随访,随访截至2022年12月31日。
1.2 方法
1.2.1HE染色 所有标本均经10%中性福尔马林固定,常规脱水,石蜡包埋,4 μm厚连续切片,常规HE染色,镜检。
1.2.2免疫组化 采用免疫组化EnVision两步法染色,抗体BCOR(克隆号C-10)、Cyclin D1(克隆号SP4)、PAX8、SATB2、TLE1、CCNB3、BCL-6、Ki-67、p53、EMA、vimentin、CK(AE1/AE3)、WT-1、NKX2.2、NSE、CD57、desmin抗体及试剂盒,均购自福州迈新公司。
1.2.3分子检测 石蜡组织8~10 μm厚切片4~5张,置于高温灭菌的1.5 mL EP管中,使用石蜡包埋组织DNA/RNA提取试剂盒,分别采用Sanger测序和二代测序检测BCOR基因。
2 结果
2.1 临床特征3例CCSK中,男性2例,女性1例,年龄10个月~7岁,平均33个月。1例发现时即发生腹膜后多发转移及全身多发骨转移,临床初诊肾母细胞瘤,并以“长春新碱+阿霉素+放线菌素”方案术前辅助化疗2周期,3例患者均行肾癌根治术治疗。
2.2 病理诊断
2.2.1眼观 手术切除肾及肿物标本3例,均为单发病变,2例位于左侧肾,1例位于右侧肾;肿瘤最大径11~17 cm,平均(13.83±2.46) cm,2例肿瘤切面灰白、灰黄色,富有光泽,呈鱼肉样,伴有出血、囊性变;例2肿瘤切面呈五彩状,质软。
2.2.2镜检 肿瘤与周围组织分界清楚,部分区域出现扇形边缘。3例均有经典的CCSK表现,肿瘤细胞排列呈腺泡状、条索状结构,间质具有丰富、纤细的树枝状血管网(图1),细胞核呈圆形、卵圆形或梭形,部分细胞核偏位呈上皮样,核染色质细腻,核仁不明显,核分裂象易见,细胞外基质富含黏多糖,使得镜下呈“透明细胞”外观,3例均伴坏死,坏死区域分别占肿瘤的10%、70%和20%;2例部分区域间质含有大量黏液,形成黏液湖,表现为黏液型(图2);2例部分区域细胞排列密集,细胞核重叠,核分裂象增加,表现为富于细胞型(图3);例2间质内含有较多无细胞性胶原成分,伴明显玻璃样变性,类似骨样基质,表现为硬化型。本组病例经典型占100%(3/3),黏液型占66.7%(2/3),富于细胞型占66.7%(2/3),硬化型占33.3%(1/3)。
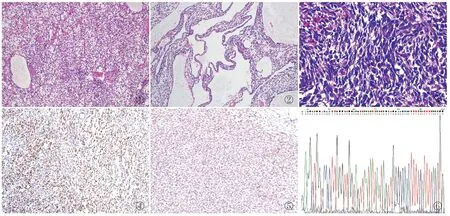
图1 肿瘤呈腺泡状结构,间质具有丰富的纤细的树枝状血管网 图2 部分区域间质含有大量黏液,形成黏液湖,表现为黏液型 图3 部分区域肿瘤细胞排列密集,表现为富于细胞型 图4 约80%肿瘤细胞核Cyclin D1阳性,EnVision法 图5 约90%肿瘤细胞核BCOR阳性,EnVision法 图6 例1 BCOR基因Sanger测序检测到BCOR点突变:c.5000C>T(p.S1667L)
2.2.3免疫表型 3例CCSK免疫组化vimentin、Cyclin D1(图4)、BCOR(图5)、TLE1均呈细胞核阳性,阳性率分别为100%(3/3)、100%(3/3)、100%(3/3)、33.3%(1/3),Ki-67增殖指数为分别为10%、30%和10%,其中例1富于细胞区域Ki-67增殖指数高达80%,p53阳性率5%~30%,平均值约11.7%,CK(AE1/AE3)、WT-1、PAX8、NSE、CD57、NKX2.2、desmin、CCNB3、SATB2、EMA、BCL-6均呈阴性。
2.2.4分子检测 例1和例2行针对BCOR基因DNA的Sanger测序检测,例1检测到BCOR点突变:c.5000C>T(p.S1667L)(图6),蛋白功能预测工具(functional analysis through hidden markov models, FATHMM)预测致病性得分0.96;例2未检测到BCOR基因改变。例3行包含BCOR基因的1 021个基因的二代测序,结果未检测到BCOR基因改变,但显示CDKN1B、CDKN2C、JUN、MCL1、SRSF2、NFKBIA、BTG1、BTG2、CALR、GATA6、CDK4、RAD21、ID3、DAXX、TERC、PTCH1、FANCF、PRKAR1A全外显子扩增。
2.3 随访3例患者均死亡,随访14~36个月,平均生存时间24个月。例1术后未行辅助化疗,随访36个月后死亡;例2术后继续沿用术前方案化疗,随访14个月后双侧脑、肝脏和腹膜后多发转移以及肋骨、脊椎、股骨等多发骨转移灶,死于多器官转移;例3术后予以“长春新碱+放线菌素”方案化疗,6个月发生双肺、左侧肾区、肝脏和腹膜后多发转移及肋骨、脊椎、股骨多发转移,随访22个月后死于多器官转移。
3 讨论
CCSK好发于1~3岁儿童,平均发病年龄36个月,男女发病比约2 ∶1[3]。CCSK发现时常为中晚期,伴有骨转移,早期也被称为“儿童时期伴有骨骼转移的肾脏肿瘤”[2],现有研究证实,脑组织已超过骨成为最常见的转移部位[4]。本组3例患者均死亡,其中例2诊断时即发生腹膜后、全身多发骨转移灶,术后发生双侧脑转移。CCSK在CT平扫呈等低为主的混杂密度,增强扫描皮质明显受压强化,瘤内血供丰富,其内富含黏液成分及坏死,呈不均匀轻-中度强化[5]。
CCSK组织学形态多样,可见经典型、黏液型、硬化型、富于细胞型、上皮样型、间变型,约91%的CCSK为经典型,经典型的结构总是伴随其他至少一种生长模式[6]。例2超过60%区域呈硬化型改变,该例患者术前行新辅助化疗,推测其形态学改变与化疗相关,其椎骨转移灶细胞形态类似于原发灶,呈经典型及硬化型。治疗后复发的病例也常显示出与原发肿瘤相同的模式,上皮样模式除外[2]。免疫组化标记vimentin肿瘤细胞核呈非特异的弥漫强阳性,Cyclin D1是敏感但不完全特异的标志物[7],BCOR抗体细胞核弥漫阳性对于诊断具有高度敏感性和特异性[8]。TLE1、SATB2、BCL-2和CD10亦显示出不同程度的非特异阳性。本组病例中vimentin、Cyclin D1、BCOR呈细胞核弥漫强阳性。
约75%的CCSK发现BCOR基因15号外显子的不同串联重复(internal tandem duplications, ITD)[9],涉及氨基酸1 700~1 755区域,受累区域几乎总是重复。本组1例检测到BCOR基因第5 000个碱基的点突变,涉及第10号外显子,本例基因改变不同于既往报道的CCSK分子特征。BCOR是位于X染色体上Xp11.4,是BCL-6相互作用共抑制因子,是一种参与控制胚胎发生、间充质干细胞功能、造血和淋巴细胞发育的转录因子,该基因存在16个外显子,编码蛋白质异构体,其中主要异构体由14个外显子编码,产生1 755个氨基酸的蛋白质[10]。BCOR蛋白具有3个已知的功能结构域,涉及第4、11及15号外显子,本例基因改变涉及10号外显子,不同于常见的功能结构域,但是该例BCOR蛋白免疫组化约90%肿瘤细胞核呈阳性,该基因的点突变可能是导致蛋白过表达的原因,这可能代表了一种新的功能区域。Cramer等[11]报道1例具有间变特征的儿童胚胎性横纹肌肉瘤中存在BCOR、ARID1A、SETD2基因突变,BCOR基因抑制组蛋白甲基化,这种活性受到组蛋白去乙酰化酶(histone deacetylases, HDAC)调节,在肿瘤多次复发后,使用HDAC抑制剂伏立诺他作为单药治疗,肿瘤存在短暂的治疗效果,表现为肿瘤坏死,患者症状改善,可停止自控镇痛;但随后肿瘤进展、破裂、穿透皮肤,患者停药后死亡。在具有相似BCOR基因改变的恶性间叶源性肿瘤中,使用HDAC抑制剂伏立诺他可能会对患者带来预料之外的治疗效果。
BCOR基因突变多发生在血液系统恶性肿瘤及恶性间叶源性肿瘤中,涉及BCOR基因的易位主要见于未分化的圆细胞肉瘤、高级别子宫内膜肉瘤、骨化纤维黏液样肿瘤,BCOR基因ITDs主要在CCSK及伴BCOR基因改变的中枢神经系统高级别神经上皮性肿瘤(central nervous system high-grade neuroepithelial tumors with BCOR alteration, CNS HGNET-BCOR)中检测到[10]。这类肿瘤具有相似的组织学及分子遗传学改变,因此Santiago等[12]推测PMMTI、CNS HGNET-BCOR及CCSK是否为一种累及软组织、肾脏和神经系统的肿瘤三联征。分子遗传学研究发现,BCOR基因改变是眼面心牙综合征(oculofacialcardiodental syndrome, OFDC)的主要原因,最常见的突变是截断和移码突变,可增强骨髓间充质干细胞的成骨/牙本质的潜能。BCOR基因突变也与骨髓增生异常综合征、小儿急性髓性白血病和慢性髓细胞白血病相关[13-14]。
在未分化圆细胞肉瘤(undifferentiated round cell sarcoma, URCS)、婴儿期原始黏液样间叶源性肿瘤(primitive myxoid mesenchymal tumor of infancy, PMMTI)、BCOR-CCNB3基因融合肉瘤和CCSK中发现BCOR mRNA高表达,导致BCOR蛋白高表达和独特的BCOR驱动转录模式,表明这些肿瘤具有高度的遗传相关性[15]。因此,Argani等[8]提出肾脏和骨/软组织的CCSK、URCS/PMMTI和BCOR-CCNB3肉瘤可被认为是具有共同遗传特征和相似临床病理学特征的肾和肾外肿瘤的“BCOR基因改变家族”。骨和软组织的BCOR-CCNB3基因融合肉瘤最初被认为是一类EWS非重排的尤因肉瘤,采用尤因肉瘤的化疗方案(多柔比星和异环磷酰胺)治疗,CCSK的治疗则强调多柔比星、不包括异环磷酰胺的方案治疗,其总体毒性较小,在CCSK中表现出明显的临床获益,CCSK的5年无瘤生存率从65%提高至85%,5年总生存率从75%提高至90%[15],若改用以CCSK为基础的化疗方案治疗BCOR-CCNB3软组织肉瘤,也许能改善患者的预后。
虽然CCSK的组织学形态较普通肉瘤良善,但其生物学行为具有侵袭性,预后较差,目前手术切除、术后联合化疗和放疗仍为主要治疗方式,因此早期正确诊断对于患者的治疗至关重要,未来针对患者的分子遗传学开发相应的靶向药物,才能进一步改善患者预后。
猜你喜欢
电子科技大学学报(2022年5期)2022-10-29 01:57:52
中国生殖健康(2020年4期)2021-01-18 02:58:10
家庭医学(下半月)(2019年11期)2020-01-16 08:39:08
家庭医学(下半月)(2019年10期)2019-11-16 08:59:52
中国临床医学影像杂志(2019年4期)2019-06-18 10:55:06
特种经济动植物(2019年9期)2019-01-08 11:35:35
数码世界(2018年1期)2018-12-23 21:39:47
中国生殖健康(2018年4期)2018-11-06 07:12:16
天然产物研究与开发(2016年6期)2016-06-05 10:29:26
郑州大学学报(医学版)(2015年1期)2015-02-27 14:50:36
