
生物质基纳滤膜制备与应用研究进展
2023-10-12 02:19宋子凡游昕达
化学工业与工程 2023年3期
宋子凡,罗 珊,陈 冉,赵 瑞,黄 彪,游昕达*
(1.福建农林大学材料工程学院,福州 350002;2.植物纤维功能材料国家林业和草原局重点实验室,福州 350002;3.鲁汶大学化学工程系,比利时 鲁汶 3001)
膜技术是基于选择渗透性膜的无相变分离技术,分离过程高效且占地面积小,相较传统热法分离技术可节省近90%能耗,因其低碳优势得到广泛关注[1]。 纳滤是分离精度介于超滤与反渗透的压力驱动膜分离技术,发展于20 世纪80 年代[2,3]。 纳滤膜作为纳滤系统的核心,孔径约0.5 ~2.0 nm,纳米级的筛分精度使其可截留相对分子质量在500 ~2 000 范围内的有机分子和多价离子[4]。 多样化的截留特性赋予纳滤技术广阔的应用场景,包括海水淡化、废水净化[5]、离子分离[6]、食品/医药产品纯化[7]和有机溶剂回收[8]等。 近年来,锂电产业的发展也推动了纳滤技术在锂离子提取等新能源相关领域的应用。
膜材料是纳滤技术的“芯片”,对于分离效率、运行稳定性、操作能耗等技术指标至关重要,进而影响纳滤过程的操作成本、投资成本等技术经济因素。 自纳滤技术面世以来,石油基聚合物材料因具有良好的成膜性与低廉的生产成本,在纳滤膜材料方面长期占据主导地位,包括聚酰胺、聚砜、聚醚砜、聚偏氟乙烯等[9]。 例如,美国陶氏公司(FILMTECTM)的标杆纳滤膜产品——NF270 和NF90 系列产品,其活性层材料即为界面聚合法制备的聚酰胺。 纵使石油基纳滤膜材料在基础研究和市场应用方面已取得长足进展,随着不可再生石油资源的过度开采,未来纳滤膜的生产极可能受此限制,而石油基聚合物加工过程的高碳排也使纳滤膜的绿色发展面临挑战。 另一方面,随着纳滤市场的逐步推广,服役到期的废弃纳滤膜也可能带来额外的环境影响。 考虑到石油基膜材料的降解难度较高,纳滤技术的全生命周期可持续性极具挑战[10]。 随着“双碳”相关政策的落实和绿色化学理念的发展,开发新型绿色纳滤膜材料已迫在眉睫。
生物质是以农林加工剩余物和废弃物为原材料,通过物理化学技术手段加工制得的可再生资源,具有“零碳排”特性。 生物质材料的种类多、分布广、来源充足。 以典型的林业生物质——木材为例,据联合国粮食及农业组织统计,全球木材年产量高达30 亿m3。 此外,生物质材料比石油基材料具有更好的生物降解性,绝大部分生物质材料在自然环境中可被微生物完全降解。 生物质材料丰富多样的功能基团可为膜制备和结构设计提供强大的化学平台,而纳滤技术广泛的使用场景也为生物质基纳滤膜的应用提供广阔舞台。 近年来,研究者已开发了包括纤维素、壳聚糖、多酚在内的多种生物质材料用于制备纳滤膜,取得了大量进展。 但是,目前膜领域对生物质基纳滤膜可控制备、结构调控与性能强化方面的认识有待加深,从生物质材料视角综述纳滤膜研究进展的工作也未见报道。本文对近年来的生物质基纳滤膜的研究工作进行简要总结,从生物质材料类型出发,围绕膜制备方法、结构调控及其对分离性能的影响逐步展开,最后总结了生物质基纳滤膜的研究进展并分析了未来的发展方向。
1 生物质基聚合物制备纳滤膜
生物质基聚合物是由生物体通过生化反应合成的天然大分子,其具有与石油基聚合物相似的高成膜性,在膜领域得到广泛关注。 例如,最早工业应用的反渗透膜即由醋酸纤维素制得[11]。 在纳滤领域,研究最多的生物质基聚合物主要包括纤维素和壳聚糖及其衍生物,前者主要源于植物而后者主要储存于海洋节肢动物的甲壳中。
1.1 纤维素基纳滤膜
木质纤维素是储量最丰富的生物质资源之一,全球纤维素年产量可达750 亿t[12]。 纤维素由重复的糖单元组成,是一种线性大分子多糖[图1(a)],分子式为(C6H10O5)n。 纤维素富含羟基使其极易形成分子内和分子间氢键,在常见溶剂中均难溶解,因而无法直接采用石油基聚合物膜的加工手段制备纤维素膜。 但羟基的高反应活性也为后续衍生化提供了便利,现已发展出包括酯化、醚化等改性方法[13],赋予纤维素额外的功能与优异的溶解性,为纤维素基纳滤膜的设计加工提供了可能。

图1 (a)纤维素化学结构;(b)醋酸纤维素化学结构;(c)壳聚糖化学结构Fig.1 Chemical structures of (a) cellulose; (b) cellulose acetate and (c) chitosan
纤维素衍生物中,醋酸纤维素的商业化程度高、应用范围广。 醋酸纤维素利用纤维素分子中的羟基与醋酸的酯化反应制备,乙酰基的引入减弱了分子间氢键并增大分子间距,进而提升纤维素链在溶剂中的溶解性和成膜性[图1(b)],在纳滤领域得到大量研究。 周金盛等通过相转化法制备醋酸纤维素(CA)-三醋酸纤维素(CTA)非对称平板纳滤膜,对NaCl 和Na2SO4的截留率最高可达60%和98%[14]。 为提升CA 膜的分离效率,Ghaemi 等在CA 膜制备过程中添加了十二烷基硫酸钠,显著减薄了膜分离层厚度并提升水通量[15]。 纳滤实验表明,CA 膜对对硝基苯酚(PNP)和3,5-二硝基水杨酸(DNSA)的截留性能受pH 值影响较大。 pH 值对DNSA 截留率的影响顺序为8.0>7.0 >5.5>4.5,而对于PNP 截留率的影响顺序为8.0>4.5>5.5>7.0。这是由于在中性pH 值条件下,PNPs 分子中的羟基发生去质子化作用,PNPs 之间形成键的几率降低,聚集体尺寸减小,更容易渗透过膜。 从荷电性来看,PNP 分子的正电性增加,然而膜的表面带负电荷,因此更易在膜表面吸附,强化浓差极化现象,导致截留率降低。 上述策略在纤维素基纳滤膜材料优化方面均取得一定效果,但采用传统试差法筛选最优膜的效率较低。 基于此,Odena 等通过遗传算法和高通量实验来加速筛选并优化CTA 纳滤膜的水处理性能[16]。 在4 代优化过程中,纳滤性能逐步提升。 这种定向搜索算法可快速筛选出截留率接近目标值的纳滤膜,对布洛芬的截留率高达96%。
除膜材料优化外,部分研究者对纤维素基纳滤膜的结构形式也进行了探索。 中空纤维膜组件相对于平板膜具有更高的装填密度和有效膜面积,在工业分离中更占优势。 于品早等基于前期报道的CTA 中空纤维反渗透膜制备技术[17],对纺丝配方、成膜条件以及后处理的工艺进行了改进,研制出性能稳定的CTA 中空纤维纳滤膜[18]。 纺制的CTA 中空纤维纳滤膜性能稳定,在1.0 MPa 操作压力下对MgSO4的截留率大于96%而对NaCl 的截留率小于50%,水通量约250 L·m-2·h-1·MPa-1。 钟蔚等用CTA 和N-甲基吡咯烷酮制备铸膜液并使用纺丝机纺制中空纤维素膜[19]。 所制得的CTA 中空纤维纳滤膜对二价盐具有较高截留性能且渗透通量受盐浓度影响较小,具有优异的分离稳定性。 此外,CTA中空纤维素纳滤膜对于维多利亚蓝、甲基橙、结晶紫和甲基蓝4 种染料的截留率均可达93%以上,尤其对结晶紫以及维多利亚蓝能实现完全截留。
制浆造纸工业的发展为纤维素的提取加工提供了技术支撑,目前已开发出了包括N-甲基吗啉-N-氧化物、离子液体、碱-脲水溶液等纤维素溶剂。上述溶剂体系可高效提取木、竹、棉等天然生物质中的纤维素并得到再生纤维素溶液,并进一步加工成膜。 此外,纤维素在多数有机溶剂中难以溶解,这赋予纳滤膜优异的溶剂稳定性,极适用于有机溶剂纳滤。 2016 年,Anokhina 等通过N-甲基吗啉氧化物(NMMO)溶解纤维素并浇铸在非织造聚酯载体上制备纤维素复合膜,首次报道了再生纤维素膜对非质子型溶剂的纳滤性能[20]。 研究发现溶剂性质对纤维素膜的选择渗透性影响较大:膜渗透性随着溶剂黏度的增加而下降,而溶质截留率则可能与溶剂对纤维素的溶胀能力有关。 研究者推测,纤维素膜溶胀程度越高则孔结构越窄,染料截留率更高,其中雷马唑亮蓝R 在四氢呋喃和二甲基亚砜溶剂中的截留率分别为42%和93%。 此后,研究人员主要在纤维素溶剂方面优化成膜过程。 Anokhina 等采用1-乙基-3-甲基咪唑醋酸盐([EMIM]OAc)混合二甲基亚砜(DMSO)溶解纤维素并制备纳滤膜[21]。DMSO 的加入可降低纤维素溶解温度并降低溶液黏度,随着DMSO 浓度的增加,纤维素溶解时间缩短,当DMSO 与[EMIM]OAc 为1 ∶1 时溶解时间最短。随着铸膜液中离子液体的浓度增加,复合膜的纤维素层形成了更致密的多孔结构,使阴离子染料橙黄II(350 Da)和雷马唑亮蓝R(626 Da)的截留率分别由21%和42%增加至42%和62%。 Sukma 等在[EMIM]OAc 溶剂体系基础上,另引入丙酮作为挥发性助溶剂制备再生纤维素膜[22]。 通过蒸发挥发性共溶剂可提高铸膜液中纤维素的浓度,进而增加膜结构致密度,使膜对溴百里酚蓝(624 Da)的截留率由 69.8% 提升至 94.0%, 但渗透速率也由84 L·m-2·h-1·MPa-1大幅降低至3 L·m-2·h-1·MPa-1。上述探索表明再生纤维素膜微结构的精密调控对提升纳滤性能至关重要。 基于此,Szekely 等使用[EMIM]OAc 溶剂体系溶解纤维素的同时,引入壳聚糖作为第2 组分调控[23]。 纤维素与壳聚糖的强相互作用使膜结构致密精密可调。 通过调节壳聚糖含量在10%~25%(质量分数,下同),所制备的膜在相对分子质量为413 ~499 Da 的范围内均具有优异截留性能。 其中,壳聚糖含量为25%的再生纤维素膜展现出稳定的乙腈渗透率,并在7 d 错流过滤中对氯沙坦和欧罗佩因的截留率分别维持在92.9%和99.9%。 此外,再生纤维素纳滤膜可在酶催化辅助下降解并在14 d 内可降解完全,具有全生命周期循环潜力。
1.2 壳聚糖基纳滤膜
壳聚糖是甲壳素脱乙酰基反应制得的生物质聚合物,与纤维素的主要结构区别在于含有丰富的氨基[图1(c)]。 壳聚糖的原料甲壳素广泛存在于海洋节肢动物等生物的甲壳中,年合成量在百亿吨规模,仅次于纤维素。 壳聚糖及其衍生物相比于纤维素具有更好的水溶性,能够以水为溶剂直接浇铸成膜,但也需通过化学交联形成稳定网络结构。 苗晶等以聚砜超滤膜为基膜,以壳聚糖硫酸酯水溶液作为铸膜液,并通过戊二醛交联制备壳聚糖硫酸酯/聚砜复合纳滤膜[24]。 复合膜具有优异脱盐性能,对Na2SO4和NaCl 的截留率分别为91.2%和48.5%。 Boricha 等制备了含羧酸基的壳聚糖衍生物N,O-羧甲基壳聚糖(NOCC)并同样以戊二醛溶液为交联剂制备复合纳滤膜[25],对水中不同浓度硫酸镍和氯化镍的最大截留率分别可达到80%和62%(5×10-6),78%和59%(10×10-6),74%和57%(50×10-6)。 为进一步提升壳聚糖膜交联度,张浩勤等直接以壳聚糖和均苯三甲酰氯分别作为水相和油相单体,界面聚合交联制备了纳滤膜[26]。 壳聚糖的氨基可与均苯三甲酰氯的酰氯基发生酰化反应,形成聚酰胺网络。 得到的壳聚糖基聚酰胺纳滤膜对聚乙二醇2000 的截留率达92%,脱盐率为7.8%~32.8%。 除了分离层交联结构外,支撑层与分离层间的交联结合对壳聚糖膜性能也有影响。Lee 等通过在聚醚砜邻位醚位引入硝基并还原生成胺基,设计含氨基功能化支撑层以增强其与活性层的结合力[27]。 随着支撑层中氨基含量的增加,复合膜表现出优异的脱盐率,对硫酸镁的截留率与商售纳滤膜几乎相同(~97.5%),而水通量是后者的1.4倍。 值得注意的是,壳聚糖纳滤膜虽具有生物降解性,但也更易遭受微生物污染。 陈国华等用浸涂壳聚糖硫酸酯溶液制备复合纳滤膜,考察了水溶性油溶性交联剂对膜性能的影响[28]。 交联后的膜表面含有少量游离氨基,赋予抗菌效果,不易附着藻类和微生物,有效提高了膜使用寿命。
纳滤分离过程除了尺寸筛分机制外,静电作用也对荷电溶质的分离也具有较大贡献。 壳聚糖含有大量氨基,可在一定pH 值范围内可赋予纳滤膜荷电性,这为壳聚糖纳滤膜的设计与应用提供了更多可能。 Zhang 等利用壳聚糖膜上的质子化铵基团(—NH+3),在pH 值低于氨基酸等电点的条件下利用静电作用截留c-氨基丁酸[29]。 在pH 值为4.69的氨基丁酸/醋酸钠混合液中,壳聚糖纳滤膜对前者的截留率为95%而对后者的渗透率达90%以上,展现出优异的分子选择性。 Miao 等以壳聚糖的两性衍生物磺化壳聚糖为活性层材料制备了一种两性离子壳聚糖纳滤膜[30]。 两性离子具有pH 值响应特性,因而膜表面荷电性能依赖于料液pH 值,进而影响脱盐率。 因此可根据不同分离任务调整溶液pH 值,提高分离效率。 为进一步增强壳聚糖膜荷电性,Huang 等以壳聚糖季铵盐为活性层,二异氰酸酯为交联剂,制备了一种正电复合纳滤膜[31]。 由于正电季铵基团(—NH+4)不受溶液pH 值影响,所制备的膜的截留相对分子质量约为560 Da,纯水渗透率为106 L·m-2·h-1·MPa-1。 除了直接改性策略,通过铸膜液共混引入其他功能材料也是提升膜性能的有效手段。 Mu 等在壳聚糖膜内引入接枝介晶化合物构建界面传质通道[32]。 杂化膜在0.4 MPa压力下通量高达2 543.3 L·m-2·h-1·MPa-1,对 NaCl的截留率仍维持在66.3%。 考虑到材料界面相容性,Weng 等选择引入同为生物质的纤维素制备纤维素/壳聚糖膜,并对共混膜进行了水解和羧甲基化改性[33],制得的共混膜对NaCl、Na2SO4和MgSO4的截留率分别为约为30%、65%和65%,对甲基橙和甲基蓝的截留率均在90%以上。 陈慧娟等进一步考察了纤维素/壳聚糖纳滤膜的染料脱盐性能,发现对刚果红的截留率为 99.99% 而对 NaCl 和Na2SO4的渗透率为90%和99%,展现出较高的染料/盐选择性[34]。 表1 总结了部分生物质基聚合物纳滤膜分离性能参数。
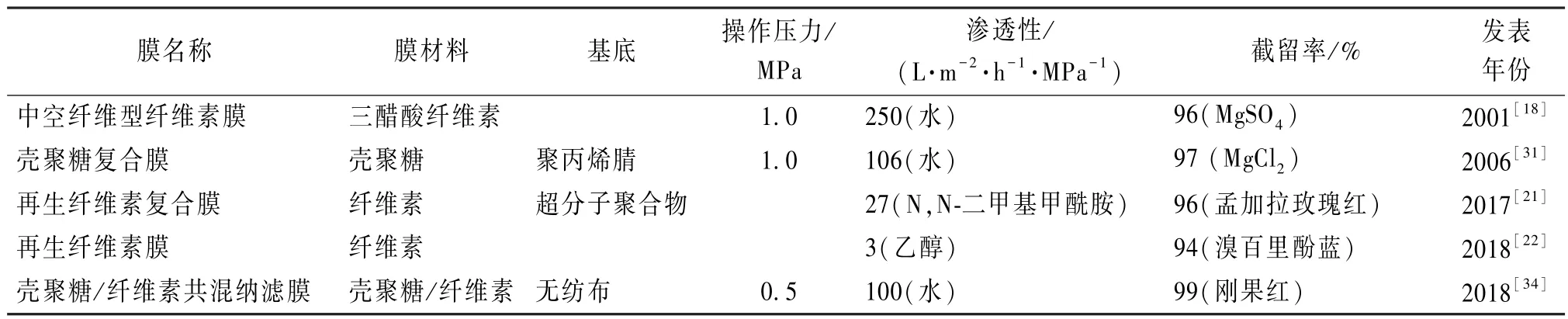
表1 生物质基聚合物制备的纳滤膜分离性能参数Table 1 Performance parameters of nanofiltration membranes made from biomass-based polymers
2 生物质小分子制备纳滤膜
除天然高分子外,自然界生物体还合成了种类丰富的有机小分子,例如多酚、植酸、寡糖等。 与生物质高分子类似,上述生物质小分子也具有大量含氧官能团,而更多样的化学结构(例如苯环、多元环等)使含氧官能团的性质及功能都发生了变化。 此外,较小的构筑单元有利于从分子尺度控制膜形成过程,实现膜结构的精密构筑和性能提升,在纳滤领域得到大量研究和应用。
2.1 多酚基纳滤膜
多酚是广泛存在于各类植物中的天然苯酚类小分子,又被称为单宁。 不同的植物来源也赋予多酚多样的化学结构,最具代表性的就是茶多酚。 我国是全球最大的茶多酚生产国,2020 年产量约5 700 万t,占全球份额的62%。 在多酚化学结构内,酚羟基具有较高的化学活性,而苯环又具有高密度的电子结构,可形成丰富的共价/非共价相互作用。 2013 年,Caruso 等在Science 上首次报道了多酚与金属离子一步配位组装策略,在水溶液中成功制得了纳米级薄膜和微囊,为多酚在纳滤领域的研究和应用拉开了序幕[35],现已演变为重要的生物质基小分子纳滤膜材料平台。
2015 年, Fan 等以单宁酸(TA) 和氯化铁(FeCl3)作为原料,通过TA 与铁离子(Fe3+)在聚醚砜基底上发生配位反应,首次制备了金属-多酚复合纳滤膜[图2(a)][36]。 研究发现随着TA 配体浓度的增加,膜截留率增加而水通量降低,可能是由于膜厚度增加导致。 在24 h 连续纳滤实验中,复合膜水通量和染料截留率均保持稳定值(下降幅度小于4.4%)。 You 等进一步研究了金属离子对多酚配位纳滤膜的影响,将聚酰亚胺基底浸泡在TA 和不同金属离子(Ag+、Co2+、Ni2+、Cu2+和Fe3+)的混合水溶液中制备了金属-多酚配位薄膜[37]。 对比发现,Co2+-TA 和Ni2+-TA 膜的渗透系数是其他配位膜的3 ~4 倍。 其中, Ni2+-TA 配位膜的接触角仅为27.5°,水通量高达456 L·m-2·h-1·MPa-1且对甲基蓝的截留率为94.1%。 相较于传统的有机相制备方法,水相配位组装条件温和,具有绿色便捷的优势。 Chakrabarty 等尝试采用乙醇作为溶剂进行配位组装,使用Cu(CH3COO)2和TA 的乙醇溶液为原料,在室温下反应形成配位网络[38]。 复合膜用乙醇清洗去除其表面的多余配合物颗粒,室温下烘干得到Cu2+-TA 复合纳滤膜,截留相对分子质量约为600 Da。
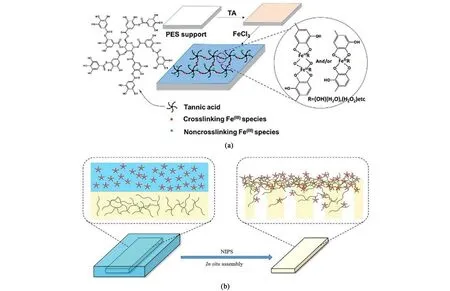
图2 (a)单宁酸-铁复合纳滤膜制备示意图[36];(b)氢键组装制备一体化多酚基纳滤膜[45]Fig.2 (a) Preparation of tannic acid-iron nanofiltration membrane[36]; (b) Synthesis of polyphenol-based nanofiltration membrane by hydrogen bonding assembly[45]
前述研究中多酚的配位或共沉积组装均发生在均相溶剂体系中,膜形成过程的可控性受溶剂产物影响,为膜结构精密调控带来不利因素。 Zhang等提出非均相界面组装的策略,实现了多酚基纳滤膜的精密构筑[43]。 单宁酸水溶液(水相)和均苯三甲酰氯己烷溶液(油相)可在多孔基底表面形成稳定界面,使单宁酸和三甲酰氯在水/油界面处接触反应生成聚酯薄膜。 由于聚合反应被严格限制在相界面处,膜结构超薄且精密可控。 此过程中可继续利用多酚的配位特性,在油相中引入钛酸四丁酯与水相单宁酸反应。 膜内的钛酸丁酯水解后形成二氧化钛纳米颗粒,构筑有机-无机界面纳米通道,随着钛酸四丁酯含量由0.02%增加到0.30%(质量分数,下同),纯水通量由668.7 L·m-2·h-1·MPa-1增加到2 047 L·m-2·h-1·MPa-1,对Na2SO4和NaCl 的盐脱率分别维持在约20%和5%,对染料截留率变化不大。 除单宁酸外,研究者还致力于探索其他类型多酚材料。 近期,Zhao 等利用槲皮素与均苯三甲酰氯的界面聚合反应制备了聚酯纳滤膜用于高盐度纺织废水的分级处理[44]。 复合膜具有39 nm 的超薄分离层,展现出良好的亲水性和负电性。 最优膜具有高渗透通量(1 980 L·m-2·h-1·MPa-1)和高选择性(极低的盐截留率为1.6%,高的刚果红截留率为99.2%)。 与商业NF270 膜相比,槲皮素基聚酯纳滤膜对染料/盐的分离效率提高288%,用水量减少42.9%。 上述多酚基纳滤膜多以复合膜的形式制备,制备过程包含多孔支撑层的相转化制备和多酚基分离层的组装制备,步骤较繁琐且面临层间剥离的风险。 为简化制膜过程,增强层间结合力,Zhang 等提出了多酚的氢键组装策略,一步法制备了一体化纳滤膜[45]。 在铸膜液内引入聚乙烯吡咯烷酮同时作为致孔剂与氢键受体,在凝固浴中引入单宁酸作为氢键供体,相转化过程二者在膜皮层通过氢键作用交联形成致密网络 [图2(b)]。 超薄无缺陷的分离层赋予了较高的水通量(152.9 L·m-2·h-1·MPa-1)和染料截留率,单宁酸亲水性则赋予优异的抗污染性能。 多酚材料作为重要的纳滤膜制备平台仍不断发展,但也面临两大挑战。 一方面,多酚复杂且难以确定的分子结构限制了膜结构的精密构筑。 另一方面,多酚配位组装体的弱稳定性也限制了膜的使用场景和寿命。 因此,兼具高组装精度和强稳定性的新一代生物质小分子材料平台亟待开发。
2.2 植酸基纳滤膜
植酸是广泛存在于植物种子的天然有机磷酸分子,又名肌醇六磷酸,化学式为C6H18O24P6。 磷酸基团比多酚羟基具有更高的给电子能力,属于强电子供体,能与过渡金属离子形成强度接近共价键的配位作用。 此外,单个磷酸分子具有6 个磷酸配位基团,配体密度极高,具有很强的螯合作用和抗氧化性,已被广泛用于食品加工、金属防腐等工业领域。 You 等首次将植酸这一天然强电子供体与金属离子组装成金属-有机磷膜(MOPM)[图3(a)][46]。通过选择具有不同离子势的配位金属离子(Ag+,Zn2+,Ni2+,Fe3+,Zr4+),可精密调控磷酸-金属配位键的键能。 对比发现,具有较高结合能或电离势的金属离子,如Fe3+和Zr4+,可以促进形成无缺陷膜结构,其中MOPM-Fe3+膜具有超亲水性,有利于水浸润和渗透。 通过改变植酸配体浓度可将MOPMFe3+膜厚度减小至8 nm,并通过改变Fe3+的含量来调节膜孔结构。 优化的MOPM-Fe3+膜具有较高的透水率(1 098 L·m-2·h-1·MPa-1)和染料截留率大于(95%)。 值得注意的是,高渗透速率将驱动更多污染物到达膜表面,抗污染问题亟需解决。 在前期基础上,Yu 和You 等通过植酸与Fe3+离子的配位作用,在多孔基底上构建了富含Fe3+离子的金属-植酸层,并通过表面Fe3+离子的桥联作用组装了全氟磺酸聚合物配体形成金属桥接氟化膜(MBFM)[47]。由于Fe3+离子的灵活调控和全氟磺酸聚合物的定向自组装,厚度仅为58 nm 的MBFM 膜具有超高的表面氟摩尔分数(47.6%) 和水下超疏油能力( ~160°),展现出1 252 L·m-2·h-1·MPa-1的高通量(比文献报道值高2 ~10 倍,染料截留率大于90%),并且对铺展和非铺展污染物型均具有强抵御能力(通量衰减率低于8%)。 但由于磺酸与Fe3+离子配位作用较弱,MBFM 膜的酸碱稳定性并不理想。 为进一步增强含氟膜稳定性,受沙塔蠕虫静电络合黏结剂启发,该团队将植酸、季铵化纤维素和全氟磺酸先后组装到多孔基底得到静电络合氟化膜(ECFMs),其中季铵化纤维素作为桥联聚电解质驱动静电络合[48]。 静电络合组装得到了厚度仅为84 nm 的氟化膜,具有精密可控的物理化学结构、表面能以及与污染物的空间相互作用。 ECFM 的透水率达930 L·m-2·h-1·MPa-1,对有机染料(>450 Da)的去除效率可达90.0%以上,可有效抵御铺展和非铺展型污染物(通量衰减率4.6%~6.7%)。 此外,强静电络合作用赋予膜优异的物理化学稳定性,ECFM 膜在长周期运行和酸性(pH=2)/碱性(pH=12)条件下均可维持高分离效率。
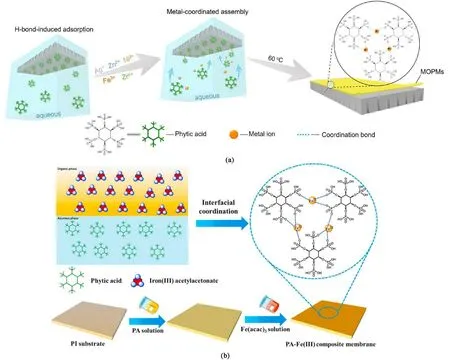
图3 (a)金属-植酸配位膜的全水相组装示意图[46];(b)金属-植酸配位膜界面组装示意图[49]Fig.3 (a) Schematic diagram of the whole aqueous phase assembly of the metal-phytate coordination membrane[46];(b) Schematic diagram of metal-phytate coordination membrane interface assembly[49]
上述工作通过静态自组装实现了植酸基纳滤膜结构的精密调控,但耗时较长。 为提升膜制备效率,Wang 等开发了一种动态选择性配位组装的方法制备了中空纤维植酸-铁配位复合膜[50]。 首先对聚酰亚胺中空纤维基底进行碱过滤改性,为后续配位组装提供活性位点。 然后,采用逐次过滤铁离子和植酸溶液的方式,在基底上动态构建植酸-铁活性层。该过滤驱动组装方法可定点修补分离层缺陷,赋予膜高渗透选择性。 与静态组装膜相比,动态组装膜的纯水渗透率从4.1 提高到85 L·m-2·h-1·MPa-1和Na2SO4截留率从68.3%提高到97.0%,远高于前期报道的静态组装植酸基纳滤膜。 此外,植酸基中空纤维纳滤膜对多种药物分子表现出很高的截留率(>99%),可高效去除地下水中的抗生素。 为进一步提升植酸的配位组装精度,Yang 等提出了植酸的界面配位组装策略,构建了具有高透过率和良好稳定性的植酸盐基纳滤膜[49]。 选择乙酰丙酮铁作为有机相金属离子来源,有效避免了传统的水相方法Fe3+的聚集和团聚[图(3b)]。 通过优化配位金属离子类型、反应物浓度及反应时间,可精密调节植酸-铁配位膜的微结构,膜厚度仅为13.4 nm,进而获得190 L·m-2·h-1的高渗透通量和99.6%的优异染料截留率。 与多酚相比,植酸在纳滤膜材料方面的研究还处于起步阶段,但其高密度的磷酸基团和规整的分子结构已吸引大量研究者开发植酸基纳滤膜材料。 充分利用植酸的物理化学特性,有望发展出新一代生物质基小分子膜材料平台。
2.3 糖基纳滤膜
小分子糖类在生物体合成代谢和分解代谢过程中都占据重要地位,是生命系统重要的能量来源。 它们一般具有羟醛或多羟酮等含氧官能团,能与活性分子发生化学反应并组装成膜。 Li 等以均苯三甲酰氯和D-葡萄糖为单体,在乙二胺交联的聚乙二胺基膜膜上,通过2 步界面聚合法制备了聚酰胺-共酯复合纳滤膜[51],水通量为425 L·m-2·h-1·MPa-1,对葡萄糖的截留率为93.8%。 Zheng 等同样采用界面聚合法制备葡萄糖基聚酯纳滤膜。 他们利用氢氧化钠加速了酰氯基与低活性羟基之间的反应,进而形成致密的聚酯网络[52]。 所制备的糖基聚酯膜水渗透率为337 L·m-2·h-1·MPa-1,Na2SO4截留率达95%,综合脱盐性能超过商业聚酰胺纳滤膜。为避免碱性试剂的使用,Shen 等开发了一种热促界面聚合方法来制备具有高脱盐性能的聚酯复合膜纳滤(图4)[53]。 在界面聚合过程中,高温促进了葡萄糖分子在水/正庚烷界面的扩散,加快了葡萄糖与均苯三甲酰氯的交联速度,进而形成比常温界面聚合方法更高交联度的聚酯膜。 最优化的聚酯膜对Na2SO4的离子截留率高达99. 5%,水透过率为161 L·m-2·h-1·MPa-1。 此外,具有高交联度的聚酯纳滤膜展现出优异的耐氯性能,在次氯酸钠溶液(1 000×10-6)中暴露36 d 后仍可保持稳定脱盐效率。
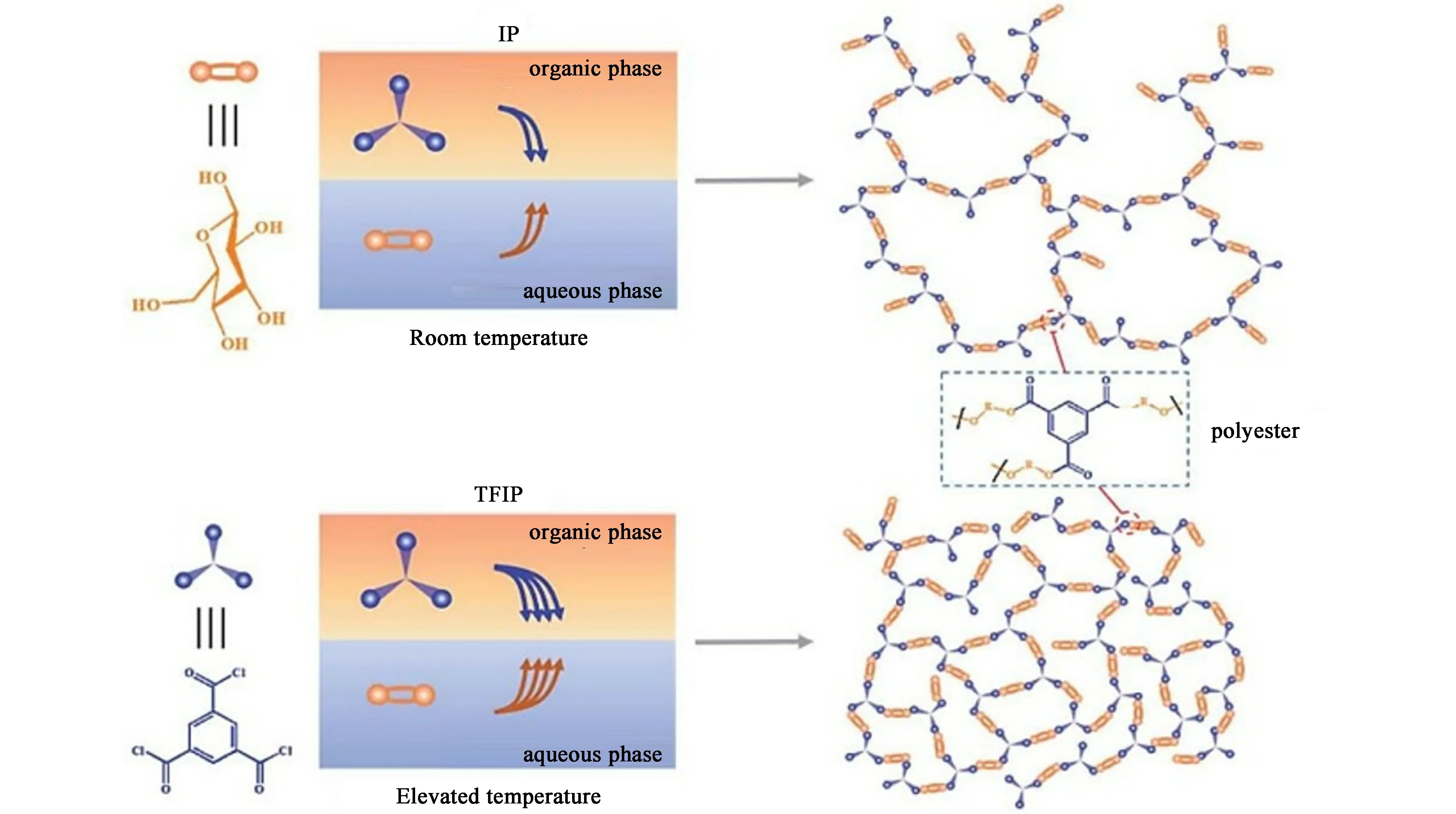
图4 热促进界面聚合制备糖基聚酯纳滤膜[53]Fig.4 Thermal facilitated interfacial polymerization for sugar-based polyester nanofiltration membrane[53]
环糊精是一类环状寡聚糖,目前研究最多的分别是含有6、7 和8 个葡萄糖单元的α、β和γ环糊精。 这3 种环糊精具有规整的空腔结构,其外沿亲水而内部疏水。 该结构与生物酶类似,可与特定分子形成主客体相互作用,在超分子化学领域已得到深入研究,该特殊性质使其在纳滤膜领域也得到广泛关注。 Villalobos 等将β-环糊精溶于水相而对苯二甲酰氯溶于有机相,采用界面聚合的方法在多孔膜上制备了β-环糊精自支撑薄膜[图5(a)][54]。 将薄膜转移至多孔基底上,即可制得复合纳滤膜。 通过改变水相的碱性,可显著提高水和甲醇通量,分别可以达到200 和94 L·m-2·h-1·MPa-1,在水、乙腈、甲醇、乙醇和四氢呋喃等多种溶剂中的甲基橙的截留率为90%或更高。 该工作首次论证了环糊精通过界面聚合制备纳滤膜的可能性,但超薄自支撑膜的转移至基底表面的难度较大,容易破损且结合力弱,不利于实际应用。 基于此,Xue 等仍以β-环糊精为水相单体,均苯三甲酰氯为有机相单体,在基底表面直接界面聚合制备了环糊精聚酯复合纳滤膜[55]。 优化后的β-环糊精聚酯膜在10 000×10-6的高浓度NaClO 水溶液中浸泡96 h,仍维持稳定水通量和染料截留率,展现出优异的耐氯性。 通过将β-环糊精质量分数从0.1%增加至1.8%,膜水通量从1 046 L·m-2·h-1·MPa-1增至1 976 L·m-2·h-1·MPa-1,对刚果红和甲基蓝的截留率分别为95.6%和94.4%。 为进一步提升环糊精基纳滤膜的渗透性,Xue 等在水相引入石墨烯量子点作为添加剂,通过界面聚合法制备了β-环糊精/石墨烯量子点复合纳滤膜[56]。 由于β-环糊精与量子点间的氢键作用诱导形成了疏松膜结构,复合膜水通量高达4 747 L·m-2·h-1·MPa-1,染料截留率保持在93.0%以上。
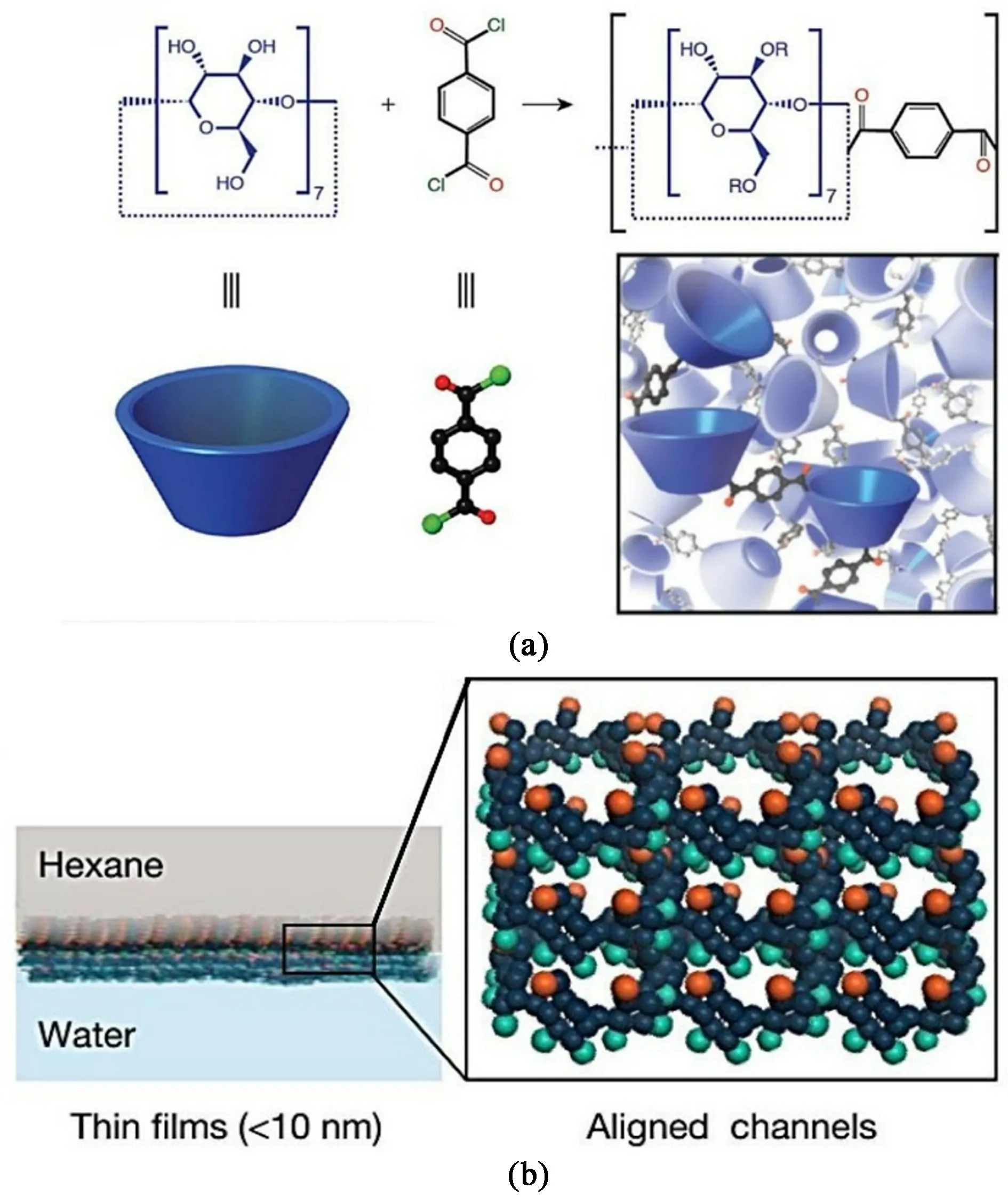
图5 (a)由β-环糊精和对苯二甲酰氯合成聚酯薄膜[54];(b)取向环糊精膜的制备过程[57]Fig.5 (a) Synthesis of mylar film from β-cyclodextrin and terephthalyl chloride[54]; (b) preparation process of oriented cyclodextrin membrane[57]
上述环糊精基纳滤膜均取得了较高的渗透通量,但并未直接利用环糊精空腔结构主导传质过程。 Liu 等通过将α-/β-/γ-环糊精和均苯三甲酰氯通过界面聚合反应生成自支撑聚酯纳米薄膜并转移至多孔基底上形成复合纳滤膜[58]。 由于环糊精内外分别形成的疏水和亲水性传质通道,3 种膜对不同极性的溶剂均具有较高渗透通量,解决了传统单一极性纳滤膜难以实现广谱高通量的难题。 3 种聚酯膜的截留相对分子质量分别为320、400 和550 Da,与测得的自由体积数据一致。 这种新型的有机分子筛膜具有可调节的孔径和清晰的孔径分布,可有效筛分不同三维尺寸的分子,其对相对分子质量较高的染料(如孟加拉红和亮蓝R)的截留率高达~99%,而对低相对分子质量染料(如甲基橙)截留率仅为86.3%。 近期,Jiang 等针对现有环糊精膜空腔结构无序排布的问题,通过界面聚合组装得到一种取向环糊精膜[图5(b)]。 他们将环糊精和4-磺基化合物[4]芳烃钠盐上部边缘的伯羟基转化为高活性的胺基,合成了氨基功能化环糊精,并将其有序排列在超薄的纳米薄膜中,创造出亚纳米级孔隙,以便在有机溶剂纳滤中精准筛分有机物[57]。 通过改变交联剂的化学性质、翻转纳米膜表面和控制纳米膜厚度来操纵大环的方向,进而操控渗透性。 研究发现,随着膜厚度由20.0 nm 减小至6.2 nm,膜结晶度显著提升,代表着环糊精单元组装的有序性增强。 通过改变环糊精种类可得到不同孔径的膜,在高附加值药物的分离方面展现了巨大的潜力。 采用不同孔径的取向环糊精膜分别去除与大麻二酚油混合的大分子以及小分子杂质,对大麻二酚油的富集率达50%,是商业膜的3 倍以上。 相比于传统生物质基纳滤膜具有的无序通道结构,该工作首次验证了生物质材料在构筑晶态取向膜方面的潜力,为生物质基纳滤膜的精密构筑提供了新参考。 表2 总结了部分生物质基小分子纳滤膜的分离性能参数。

表2 生物质基小分子制备的纳滤膜分离性能参数Table 2 Performance parameters of nanofiltration membranes made from biomass-based small molecule
3 生物质基纳滤膜的应用前景展望
综上所述,生物质作为一种可再生材料资源,在纳滤膜领域受到研究者的广泛关注。 为实现生物质基纳滤膜的精密构筑与性能强化,开发绿色、便捷、高效、通用的膜材料加工制备方法已成为研究热点。 本论文从生物质基高分子和小分子材料出发,较全面地综述了高性能生物质基纳滤膜的设计策略与制备途径,并对未来生物质基纳滤膜的发展方向作出展望。
(1)材料多样性。 目前用于制备纳滤膜的生物质材料种类相比于石油基材料仍十分有限,因此未来探索需要聚焦在新型生物质基纳滤膜材料,通过林业工程与化学化工等学科的交叉融合,充分挖掘具有制膜潜力的生物质分子平台。
(2)结构精密性。 大部分生物质分子制得的纳滤膜具有无定型结构,其通道微环境难以精密设计,导致筛分精度和效率受限。 开发新型分子自组装方法,针对分离物系特征设计精密设计膜结构,将成为未来生物质基纳滤膜的重要研究方向。
(3)制备可控性。 相比于石油基分子原料,生物质分子原料的均一性相对较差,杂质组成复杂,重点关注膜制备过程的可控性与可重复性,对于生物质基纳滤膜的放大制备与实际应用至关重要。
总之,在在“双碳”背景下,随着生物质化学化工的迅猛发展,生物质材料的加工利用水平将不断提升,结构可调、绿色低碳的生物质材料将逐步发展成为高性能纳滤膜的重要材料平台,为纳滤技术的可持续发展提供不竭动力。
猜你喜欢
中亚信息(2019年7期)2019-01-29
中成药(2018年8期)2018-08-29
中成药(2018年4期)2018-04-26
意林(环球儿童文学)(2016年12期)2016-02-20
畜牧兽医科技信息(2015年5期)2015-12-27
兽医导刊(2015年8期)2015-03-25
食品科学(2013年13期)2013-03-11
食品科学(2013年8期)2013-03-11
浙江农业科学(2012年10期)2012-12-24
动物营养学报(2011年4期)2011-03-28
