
长波长驱动光开关染料分子研究进展
2023-10-07 12:34张志伟杨伟鑫张隽佶
化工进展 2023年8期
张志伟,杨伟鑫,张隽佶
(华东理工大学化学与分子工程学院,精细化工研究所,上海 200237)
光是调节材料和生物系统的理想刺激源。日常生活中遇到的许多自然和人造系统[例如视蛋白产生视觉[1]以及光刻制造集成电路(IC)[2]]都依赖光进行功能调控[3]。在该类系统中,发色团对光子的吸收为化学过程提供了所需能量,如光异构化、光氧化或光分解。其中,可以在两种不同波长光激发下进行可逆异构化的发色团分子通常被称为光开关分子。光开关分子作为光敏单元被广泛应用各领域,利用光照后分子在具有不同化学性质的状态之间切换,可以实现对目标系统的可逆控制[4-5]。传统的光开关分子在自然光下异构化后发生显著颜色变化,已被成功应用于太阳镜和化妆品中[6]。然而,在过去的二十年里,光开关分子被应用到多学科研究中,其在分子水平上光异构体间的性质差异(例如几何和电子结构)使得精密操作复杂化学和生物化学体系成为可能。例如:利用两种光异构体之间的相互转化作为二进制信息存储手段,制备分子逻辑/存储器设备[7];利用光开关控制疏水性和亲水性,开发光控智能表界面材料[8-9];通过光照控制电导性能,制备新型有机场效应晶体管(OFET)和单分子器件等尖端半导体材料[10-11];通过影响光异构化后的分子几何形态和/或电子性能,构建手性选择性光开关催化剂,从而精细控制化学反应结果[12]。此外,光开关分子同样还被用于控制聚合物相变和储能等领域[13]。
随着生物技术的蓬勃发展,光开关分子作为光控单元可以实现对生物大分子(如多肽、蛋白、寡核苷酸和代谢物)的可逆激活/失活[14-15]。光是一种理想的生物正交和远程可控非侵入刺激,可以对特定组织甚至单个细胞中的操作进行时空控制[16]。近年来,光开关分子逐渐被应用到光药理学、光遗传学和超分辨率成像等新兴交叉前沿领域,展现出潜在临床应用前景[17-19]。因此,推动生物相容性光开关分子从基础科学走向现实应用,已成为近年来的研究重点和热点话题。
目前,构建生物相容性光开关分子迫切需要解决的问题之一是避免紫外光的使用。紫外线组织穿透力差,且对细胞具有毒害作用。然而,迄今已报道的许多经典光开关都不可避免地需要使用紫外光对光异构化的一个或两个方向进行激活(相应的异构体只在紫外区域吸收),极大程度限制了光开关分子的应用多样性。相反,可见光(λ=450~800nm)和近红外光(λ=800~2500nm)具有高组织穿透性(由于内源性血红蛋白或黑色素等光学散射和吸收)、低背景信号(避免激发生物发光)和低光毒性(避免与DNA、RNA和脂质的光反应)等优点,满足生物应用需求[20-21]。此外,对非专业用户而言,可见光/近红外光源较高能紫外光源更廉价易得。因此,开发激发能量更低、生物相容的可见光/近红外驱动光开关分子成为该领域亟待解决的关键科学问题之一。相对于传统合成的小分子光开关,可见光/近红外的仿生发色团往往需要更大的π电子体系,其设计与合成对化学家来说无疑更具挑战。
过去十年,在科学家们的不断努力尝试下,光开关分子激发波长被成功红移,新型可见光/近红外驱动的光开关分子层出不穷。本文阐述了传统光开关染料(偶氮苯、烯烃马达和二芳基乙烯)以及近年来新兴的光开关染料(靛蓝、二氢芘及给体-受体Stenhouse 加合物)在激发波长红移方面取得的重大进展,并对光转换效率(光照下异构体间相互转化的比例)、异构化量子产率(参与异构化反应的光子数与吸收光子数之比)、热力学不稳定异构体热半衰期(半数异构体在黑暗条件下转化为初始构型所需要的时间)以及抗疲劳性(光开关分子多次循环后抵抗生成副产物的能力)等光开关性能进行评价(图1)。最后,展望了可见光/近红外光开关染料分子在未来发展过程中面临的机遇和挑战。
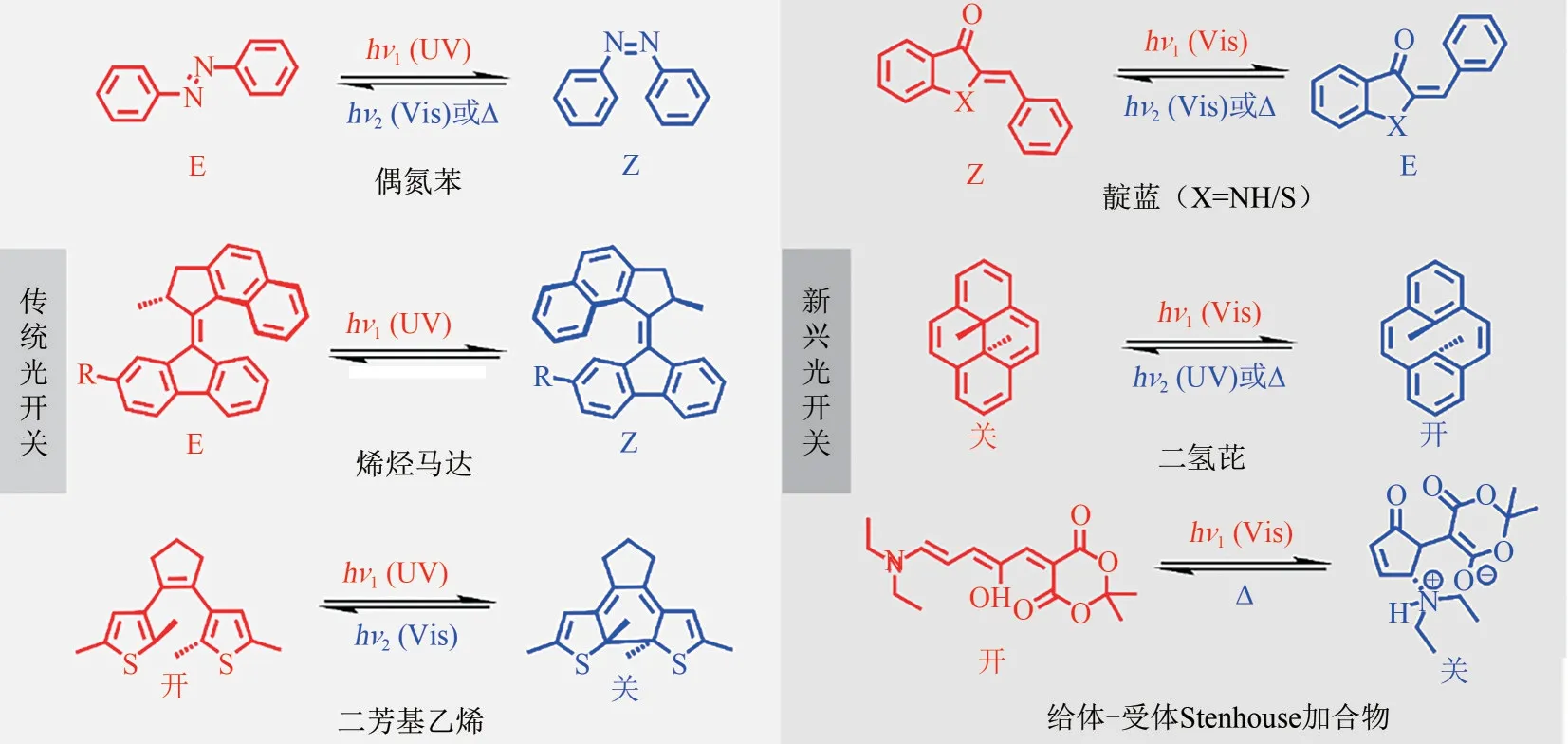
图1 常见光开关染料分子结构式以及光异构化示意图
1 传统光开关染料
1.1 偶氮苯
偶氮苯是最常见的光开关分子之一,光照条件下可以在扁平的反式构型与折叠的顺式构型之间进行反复切换。偶氮苯因其达到光稳态(特定波长光照射下构型达到平衡时的状态)时的高转化率、高量子产率以及高光稳定性等优点,展现出了巨大的应用前景。通过激发π→π*吸收带(通常为310~360nm),偶氮苯分子可以由反式构型变为顺式构型。激发n→π*吸收带(450nm左右)可以使偶氮苯恢复初始的反式构型。直接延长偶氮分子π共轭体系或在偶氮苯两端分别引入给电子基团、吸电子基团形成“推-拉”体系,均能实现偶氮苯分子激发波长的红移。值得注意的是,由于π→π*和n→π*吸收带不可避免的重叠,偶氮苯光开关性能往往受限,例如达到光稳态时转化率相对较低,这极大程度限制了偶氮苯分子的实际应用。因此,构建长波长驱动的兼顾异构化效率的偶氮苯分子显得尤为重要。
1.1.1 桥连偶氮苯
Herges 等[22-24]于2009年报道了一类桥连偶氮苯分子(AB 1-5),可以在不使用紫外光的情况下实现光异构化,如图2(a)所示。化合物AB-1[22]结构中由于桥连结构的几何张力,Z 构型与E 构型的n→π*吸收带实现了显著分离(约100nm)。因此,Z→E和E→Z的构型变化分别可以使用蓝光(400nm)和绿光(550nm)进行驱动,如图2(b)所示。环张力的存在还导致了化合物的热稳定性反转,即与传统的偶氮苯分子相比,AB-1 的Z 型异构体比E 型异构体更稳定(正己烷中半衰期τ1/2达到4.5h)。利用桥偶氮苯结构的优异性能,该课题组2016 年进一步将氧原子和硫原子引入桥接单元,得到偶氮苯AB-2 和AB-3[23]。二者呈现出类似的吸收谱图变化:光照后,n→π*吸收带由初始的400nm(Z 构型)红移至525nm(E构型),并延伸到700nm。相应地,所得E 构型经过远红光(660nm)照射可以完全转化为初始的Z构型。二者均具有良好的抗疲劳性,经多次循环操作后均未检测到吸收信号的衰减[图2(c)]。与母体分子AB-1 相比,AB-2 的半衰期大幅下降,在四氢呋喃溶液中降至89s(20℃)。相反,AB-3 呈现出较好的热稳定性,在丙酮溶液中半衰期可达3.5天(27℃)。随后,该课题组通过进一步的构效关系研究,又报道了两种近红外驱动的桥连偶氮苯AB-4 和AB-5[24]。AB-4 的最大吸收波长位于554nm附近且尾部吸收带延伸至近红外区域。使用近红外光(740nm)激发,AB-4 可以发生高效的E→Z 异构化反应,如图2(d)所示。化合物AB-5则具有较好的水溶性,光异构化反应可以在水中高效进行(水中半衰期长达1.2h),在光药理学等生物领域中具有广阔应用前景。

图2 桥连偶氮苯染料分子结构及其光开关性能
1.1.2 邻位四取代偶氮苯
在偶氮苯邻位上引入供电子基团同样可以实现Z型-E型异构体n→π*带分离[25-27]。2011年,Woolley等[25]通过在氨基偶氮苯分子邻位引入4 个甲氧基,所得化合物AB-6 的n→π*带出现了明显红移(约35nm)[图3(a)、(b)],化合物E→Z与Z→E异构化过程可以分别使用绿光(530~560nm)和蓝光(460nm)激发。当化合物以E型异构体存在时,由于O原子孤对电子与临近N 原子孤对电子之间的强相互作用,分子HOMO轨道能级提升,从而导致n→π*跃迁能量降低,实现吸收带红移且与Z 型异构体分离。此外,AB-6 的Z 型异构体在水溶液中的半衰期达到2.4 天。将化合物共价偶联到多肽结构中,AB-6 的异构化过程可以通过红光(635nm)进行高效激发,E→Z 转化率接近完全(98%),Z 型异构体半衰期达到6h,在活体应用中展现出潜在应用价值[26]。在上述工作基础上,Woolley等[27]进一步将二氧六环结构与偶氮苯苯环融合,结合现有甲氧基取代,构建了新型邻位取代偶氮苯分子AB-7。通过引入吡咯烷基与砜的结构单元,化合物在生理条件下(pH=7.2)于597nm 处形成新的长波长吸收带。AB-7可以在720nm光照下进行高效E→Z异构化反应,其Z型异构体半衰期约为1s,如图3(a)所示。
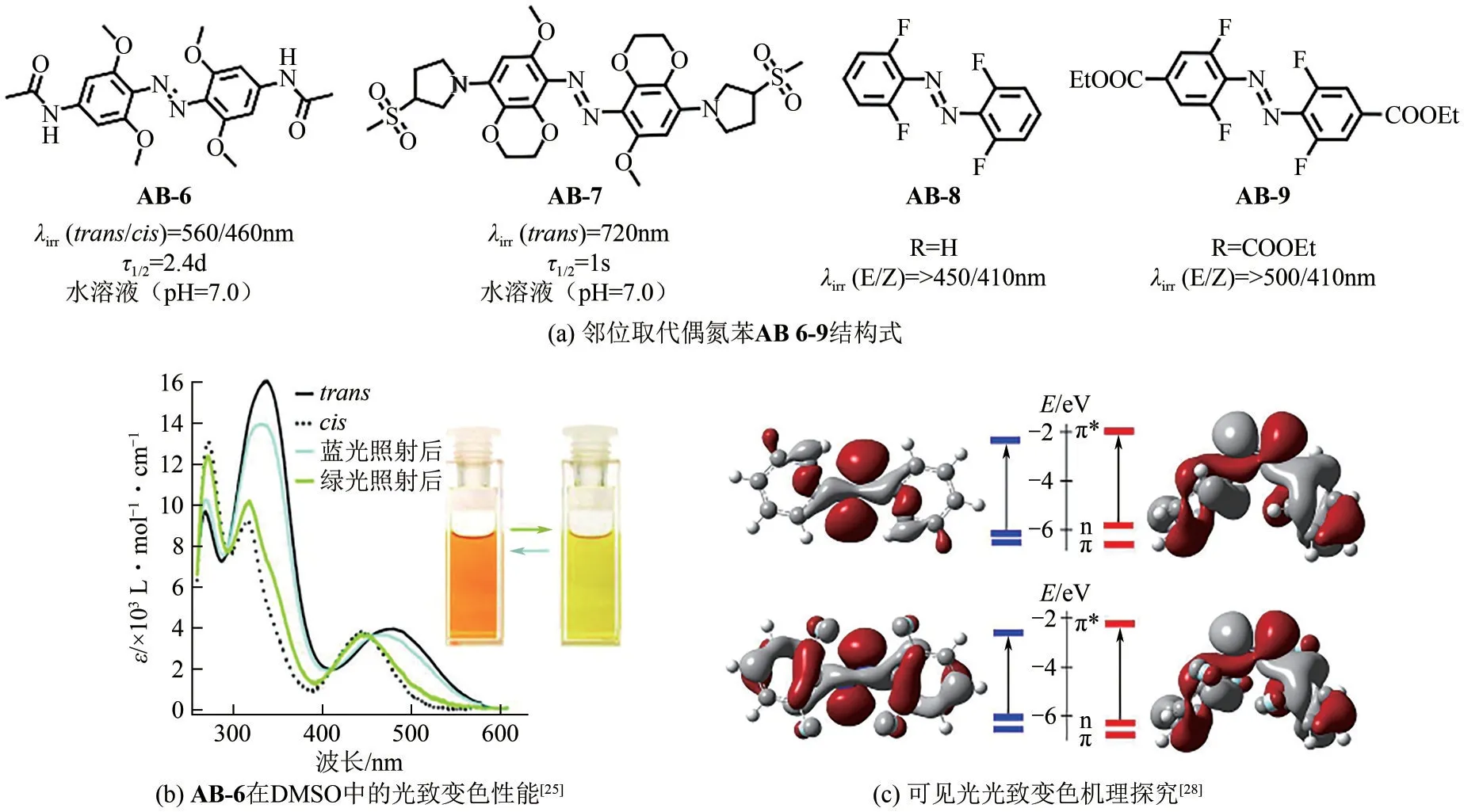
图3 邻位四取代偶氮苯染料分子结构、光开关性能及其机理探究
引入吸电子基团也可以实现偶氮苯分子吸收波长的有效分离。Hecht 等[28]设计了一系列邻位氟原子取代的偶氮苯分子[图3(a)]。如图3(c)所示,邻位引入氟原子有效减弱了N= = N 双键上电子互斥作用,Z 型异构体n 轨道能量降低,异构体间n→π*吸收带显著分离。因此,化合物AB-9 的Z 型异构体可以在>500nm光照下得到,且转化率高达90%;使用410nm的蓝光照射,Z型异构体几乎可以完全恢复到初始的E型异构体。此外,该类氟化偶氮苯分子呈现出了极佳的热稳定性。例如,化合物AB-8 的Z 型异构体半衰期长达700 天。得益于优良的性能,该类长波长偶氮苯分子被成功应用于可见光响应驱动器设计及光控MOF材料构建。
1.1.3 络合型偶氮苯
Aprahamian 等[29]报道了一种基于BF2络合的双向长波长偶氮苯分子,首次实现了偶氮苯分子的近红外光激发(图4)。与传统分子相比,该类偶氮苯分子的π→π*跃迁能量要低于n→π*跃迁,这是由于偶氮基团与BF2络合后,分子n 轨道能量显著降低,同时大共轭N—C—C—N—N 骨架导致分子π轨道能量显著提升。因此,该类分子的双向异构化过程均可以通过可见光进行激发,Z型异构体同样展现出良好的热稳定性(AB-10 的半衰期达到12.5h)。此外,该类分子的激发波长及光开关性能可以通过化学手段进行有效调控[30]。例如,在偶氮苯分子对位引入二甲胺官能团,AB-11 的吸收峰从530nm/480nm红移至680nm/620nm,且吸收带尾部可以延伸至近红外区域(760nm),异构化反应可以成功被近红外激发。然而,与未取代的AB-10相比,AB-11 异构体的半衰期从12.5h 降低至250s,同时光转化率从90%下降到63%。值得一提的是,通过控制自组装的粒径大小,该类偶氮苯分子的半衰期可以得到有效调控。
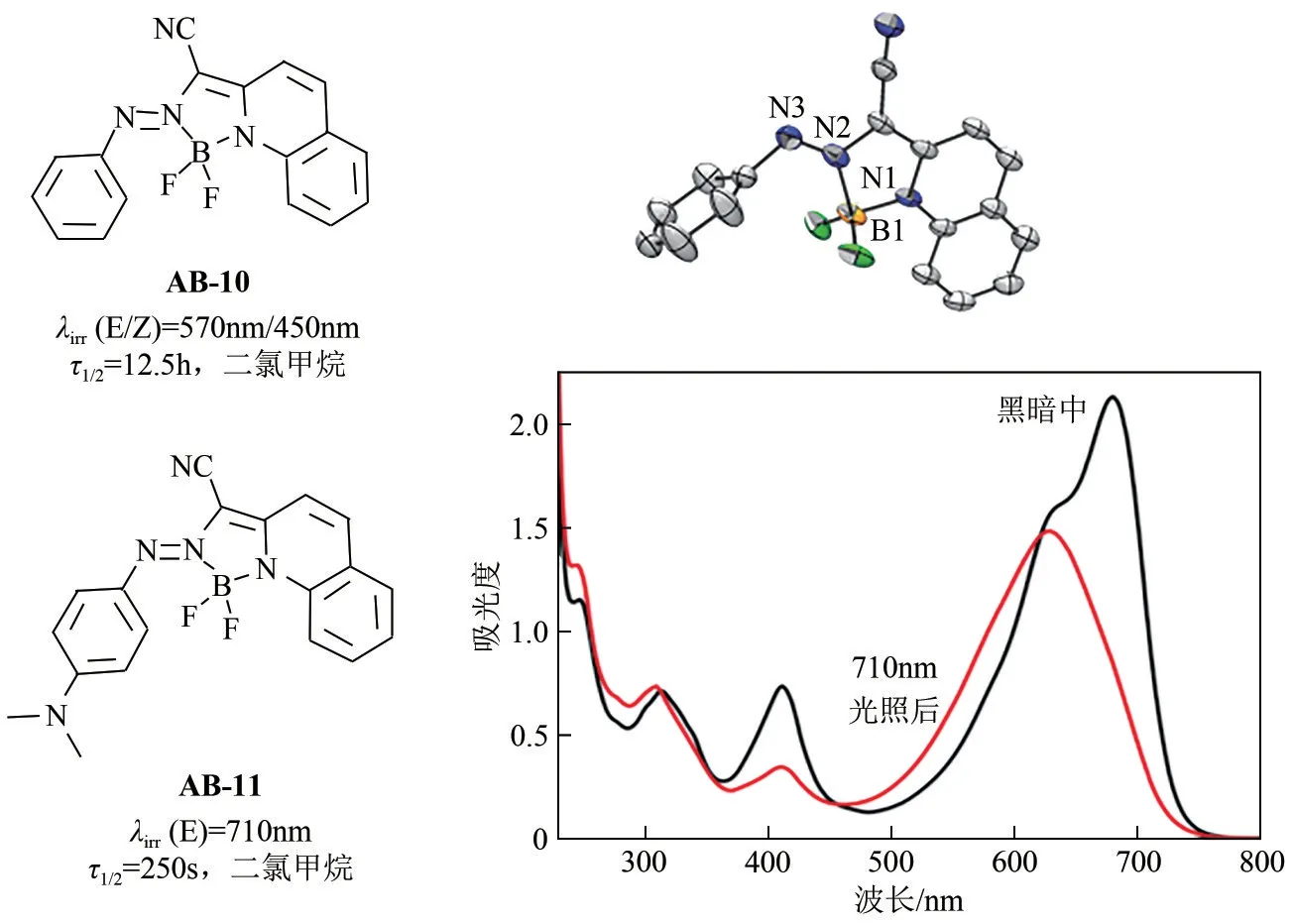
图4 BF2络合型偶氮苯染料分子式、晶体结构及其光开关性能[29-30]
1.1.4 双光子吸收
双光子吸收(TPA)是指原子或分子同时吸收两个光子而跃迁到高能级的现象,借助双光子吸收可以有效实现光开关分子吸收波长红移。1989 年以来,Rentzepis 等[31]报道了一系列基于螺旋吡啶、蒽和俘精酸酐的双光子光开关染料分子。借助双光子直接吸收过程,偶氮苯染料可以实现长波长激发,然而该种情况下其异构化性能往往大幅受限(热稳定性极差)。间接双光子吸收技术(经过分子内能量传递)则可以有效避免上述弊端,是构建双稳态双向长波长偶氮苯分子的有效策略。如图5所示,将具有双光子吸收特性的三苯胺单元与偶氮苯分子共价相连,确保三苯胺发射波长与偶氮苯n→π*吸收带重叠,所得二分体因此可以捕获低能量光子(750nm)并触发相应Z→E 异构化反应[32]。使用绿光(510nm)照射化合物AB-12,二分体又可以完全恢复到初始的Z型异构体。
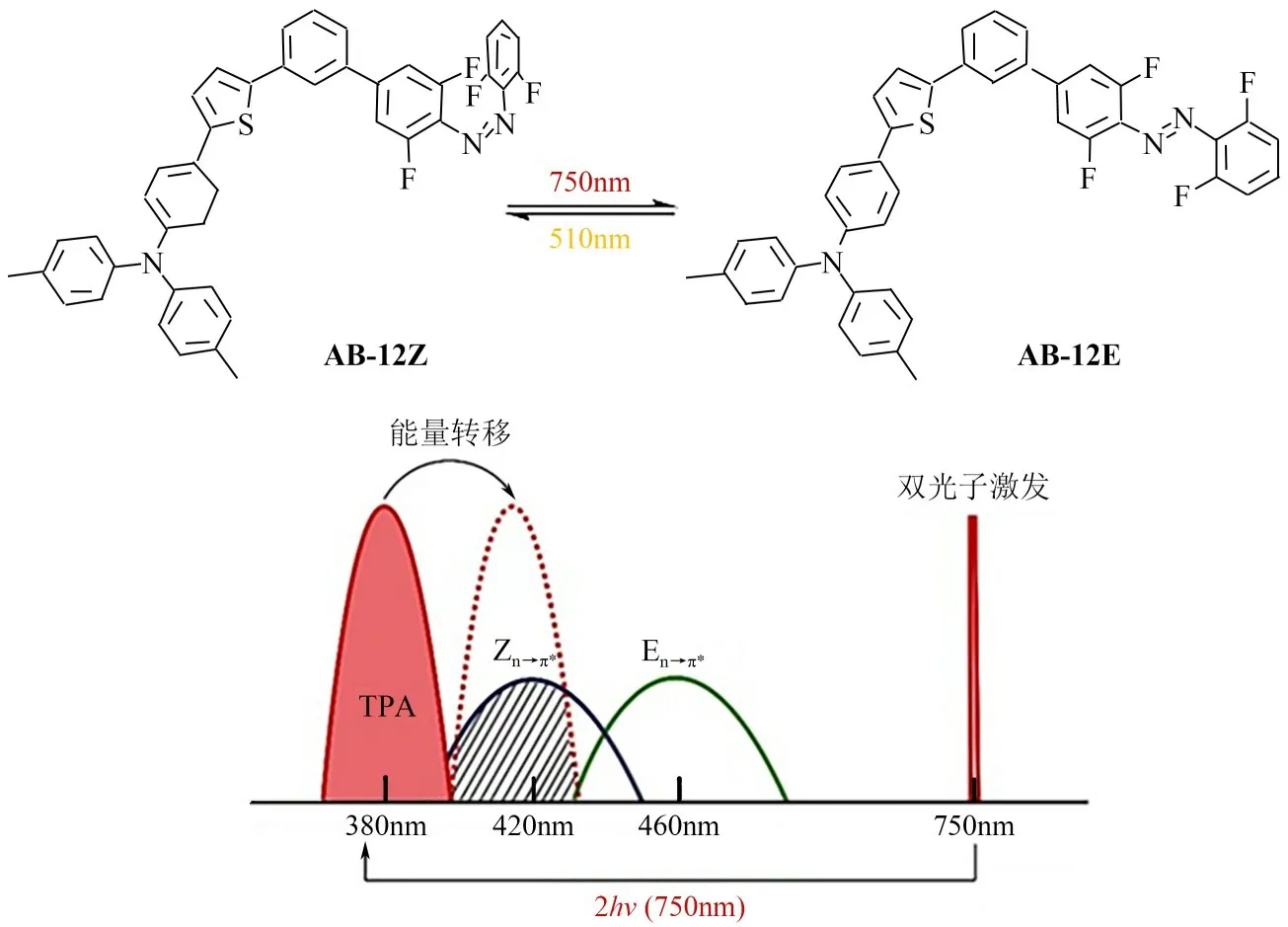
图5 双光子吸收近红外偶氮苯AB-12结构式及机理示意图[32]
1.2 位阻型烯烃马达
自从被Feringa 首次报道以来,大位阻烯烃分子马达[33]已经被广泛应用到纳米技术、催化、离子结合等领域[34]。如图1所示,分子马达经光照后生成的不稳定顺式异构体具有空间拥挤的轴向(Meax)取向,通过能量下坡热螺旋反转(THI)过程可以恢复赤道(Meeq)取向,释放空间拥挤的应变力,实现180°的单向旋转。重复上述过程即可实现完整的单向360°旋转。由于两个异构化过程具有相似的吸收带特性,因此本文主要关注分子马达的第一个光诱导E→Z异构化过程。
1.2.1 推-拉电子体系
红移分子马达最常用的方法之一是缩小分子HOMO与LUMO轨道之间的能级差。Feringa等[35]合成了一种供体-受体型功能化分子马达M-13。使用435nm光照,化合物光异构化(顺式→反式)过程可以被高效激发,转化率高达90%。加入TFA,分子马达被质子化,顺式→反式异构化速率显著提升,达到光稳态的时间由初始的720s下降至420s,同时转化率提升至95%。为了进一步缩小HOMO与LUMO 轨道能级差红移马达激发波长,Feringa等[36]分别在分子马达转子、定子部分引入了给电子、吸电子官能团,构建了“推-拉”型分子马达体系。如图6所示,将甲氧基和氰基基团引入马达分子结构,M-14 的最大吸收波长(λmax=453nm)实现了80nm 红移,化合物因此可以被530nm 绿光高效激发,其异构化量子产率达到5.84%,热不稳定异构体半衰期为1900s。高效的光异构化性能表明该策略在红移激发波长的同时并不会显著降低光反应性能。此外,该策略近期还被成功应用于构建可见光驱动的超分子金属大环体系[37]。

图6 可见光驱动分子马达M-13~M-24结构式及其性能汇总
1.2.2 共轭延伸法
增大定子或转子部分共轭程度是实现分子马达激发波长红移的另一种常用策略。如图6所示,定子部分共轭程度增加后,马达M-15的吸收峰红移至490nm[38]。可见光(420nm)光照后,马达在光稳态时转化率达76%,同时由于空间位阻的增大,马达旋转速率略有降低,热螺旋反转过程受阻。除此之外,通过增大分子马达转子部分的π 共轭体系,同样可以实现激发波长红移。将马达转子部分的萘替换为芘,马达M-16 最大吸收波长红移至414nm。虽然光异构化量子产率略低至1.4%,M-16仍然可以被蓝光(455nm)激发[39]。该策略同样可以用于构建门控型分子马达,例如引入二芳基乙烯单元后,马达分子M-17的π 共轭程度增大,可以被可见光(λirr=455nm)高效激发[40]。
改变马达定子结构也被证明是改变激发波长的可行方法。吲哚酮类分子在太阳光照射下可以发生E-Z 异构化,受该工作启发,Wezenberg 和Feringa等[41]报道了一系列由可见光驱动的吲哚酮类分子马达(图6)。化合物(M-18~M-20)的吸收波长均延伸到近500nm 处,可以被蓝光(420nm)激发,同时通过对马达转子结构微调可以有效调控马达半衰期(毫秒~几天),为构建光响应型功能材料提供分子基础。
1.2.3 能量转移法
采用三重态敏化、多光子吸收等间接方法同样可以构建长波长驱动马达。Feringa等[42]通过将分子马达与过渡金属络合,利用能量较低的金属-配体电荷转移(MLCT)带,经分子内三线态-三线态能量传递(TTET),成功实现了分子马达的蓝光激发。如图6 所示,分子马达M-21 可以与RuⅡ及联吡啶进行配位,形成的络合物最大吸收波长红移至450nm(dπ→π*跃迁)。因此,M-21 可以被蓝光(λirr=450nm)激发且旋转速度提升了近50 倍。金属-配体相互作用可以有效调节分子马达的光物理和热力学特性,为构建可见光驱动的分子马达提供了一种更简单和实用的方法。
传统的有机三线态敏化剂同样能够实现分子马达激发波长的红移。Browne 和Feringa 等[43]将具有可见光吸收特性的钯四苯基卟啉(PdTPP)敏化剂与分子马达共价相连,通过三线态-三线态能量传递过程成功实现了传统马达的高效可见光(530nm)激发。在此基础上,Browne 和Feringa 等[44]进一步利用该类敏化剂作为框架,马达分子M-23作为支柱,构建了金属有机框架(MOF)材料,实现了分子马达在固态框架材料中的高效可见光激发。
Feringa等[45]将能量匹配的双光子敏化剂与分子马达共价连接,构建了基于双光子敏化的近红外分子马达M-24。双光子敏化剂吸收近红外光子实现高能级激发,通过共振能量转移(RET)过程敏化分子马达,整个敏化过程如下:敏化剂吸收近红外(800nm)光子后S1能级被激发,能量随后传递至马达分子S1能级,分子马达进行旋转。激发态振动弛豫后,通过锥形交叉点(CI)进入基态,形成原始稳定异构体M-24s和M-24m的混合物。值得注意的是,该分子即使在极低的浓度(1.1×10-5mol/L)下仍能正常工作,有望在活体内实现共定位敏化。
1.3 二芳基乙烯
二芳基乙烯是一类基于己三烯电环化反应机理的光致变色染料分子。作为典型的p型光致变色分子,二芳基乙烯自1988 年首次报道以来便以高效的光开关效率、良好的抗疲劳性和优异的热稳定性而闻名。近年来,二芳基乙烯分子已经被广泛应用到各个领域,但可见光/近红外驱动的二芳基乙烯分子却鲜有报道。
1.3.1 侧链/烯桥共轭延伸
通过偶联芳香染料扩展二芳基乙烯分子π共轭系统,可以有效降低HOMO-LUMO 能级差,实现吸收波长红移。然而,该策略一定程度上会造成光开关性能受限。Irie 等[46]报道了迄今为止吸收波长最长的二芳基乙烯分子。通过在所选二芳基乙烯分子芳基单元2 号位引入苝单酰亚胺(PMI)或苝双酰亚胺(PDI)染料分子,光开关分子吸收波长显著红移。如图7 所示,DAE-25 和DAE-26 的最大吸收波长分别红移至543nm和554nm。在560nm的绿光照射下,二者均发生快速闭环反应,闭环量子产率分别为6.4×10-4(DAE-25)和3.1×10-3(DAE-26),同时二者开环体均几乎完全转化为闭环体(>99%)。理论计算结果表明,化合物DAE-25和DAE-26的LUMO轨道电子分布均离域到整个分子骨架,确保光环化反应的顺利发生。然而,二者的光开环反应过程却呈现出明显不同。DAE-25闭环体中分子共轭被切断,吸收波长蓝移。在405nm 的蓝光照射下,发生开环反应,分子恢复至初始状态。值得一提的是,该体系中闭环体吸收波长较开环体蓝移这一现象与传统二芳基乙烯不同,这是因为传统二芳基乙烯通常是从5,5′位置进行取代,闭环体比初始开环体拥有更大的π 共轭系统。与DAE-25 相比,DAE-26则无法进行光开环反应。理论计算和荧光激发光谱显示,DAE-25 和DAE-26 闭环体的激发态与苝酰亚胺吸收之间存在交叠,存在分子内能量转移的可行性。DAE-26光开关分子中二者的跃迁偶极矩取向几乎平行,有利于能量转移的发生,而在DAE-25中二者则呈现出垂直构型,能量转移过程受限。因此,DAE-26 闭环体激发态被快速猝灭,开环过程受到抑制,仅能单向工作。研究还发现该类分子前线轨道能量及LUMO轨道分布受多个因素影响,如噻吩环硫原子是否被氧化、取代位点(2/2′或5/5′)以及取代基的种类等,这些因素在不同程度上均会影响光开关分子性能,因此采用该策略构建长波长驱动二芳基乙烯分子往往需要精心的结构设计。
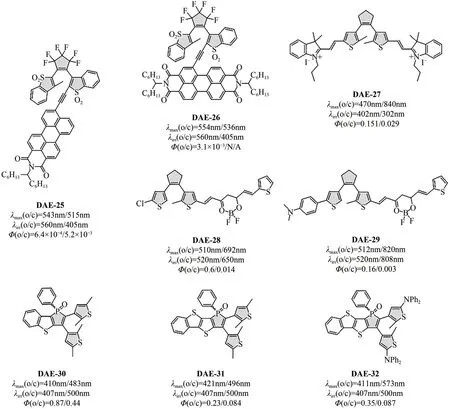
图7 共轭延伸型可见光二芳基乙烯分子结构式及其光开关性能汇总
Yin 等[47]将氰基类受体-π-受体(A-π-A)染料引入二芳基乙烯结构侧链单元,成功实现π共轭体系延伸。由于开环体在470nm 处具有强吸收特性,利用可见光(>402nm)照射,DAE-27发生高效闭环反应,闭环转化率为79%,量子产率达到0.151。尽管得到的闭环体呈现出近红外吸收带(840nm),但开环过程仅能在紫外光(302nm)照射下进行。化合物中惰性基团具有强缺电子特性,因此对CN-阴离子具有选择性响应特性,可以作为荧光探针对水溶液中的氰化物进行检测。加入氰化物后,分子初始大共轭体系断裂,DAE-27溶液颜色从黄色变为无色,荧光强度下降,化合物只能通过紫外光(302nm)诱导光闭环反应。
Guo等[48]将基于BF2络合的大共轭分子共价引入到二芳基乙烯单元的侧链上,同样实现了激发波长有效红移。受益于高强度的π→π*跃迁,DAE-28开闭环体最大吸收波长分别位于480nm(ε=8.46×104L·mol-1·cm-1)和510nm(ε=9.17×104L·mol-1·cm-1)。使用绿光(520~530nm)照射,化合物在甲苯溶液和聚甲基丙烯酸甲酯(PMMA)薄膜中都能发生高效闭环反应。吸收谱图显示所得闭环体最大吸收波长达到800nm,采用红光(650~660nm)甚至近红外光(800~810nm)照射,闭环体可以顺利发生开环反应,相应量子产率为0.014。通过对二芳基乙烯另一侧链进行修饰,可以进一步调控闭环体吸收波长。例如:引入给电子的咔唑基团后,化合物DAE-29 闭环体吸收可以红移至1100nm(最大波长为808nm)。
除了在侧链部分延伸π体系外,对二芳基乙烯烯桥部分进行π体系扩展同样可以开发可见光光开关分子。Yam等[49]报道了一系列双向可见光驱动的新型磷取代二芳基乙烯衍生物。与母体化合物相比,DAE-31 的开环体吸收红移36nm,而化合物DAE-32 的闭环体最大吸收波长则实现最大红移(中心波长位于573nm)。所有化合物均表现出良好的可见光光致变色性能,例如:DAE-30可以分别在蓝光(410nm)、绿光(500nm)的照射下进行闭环(ΦO→C=0.87)、开环(ΦC→O=0.44)反应,且在10个光致变色循环中没有明显的性能损失,呈现出良好的抗疲劳性。该类光开关分子在红移激发波长的同时其光致变色性能并未受到明显损害。
1.3.2 三线态敏化法
三线态敏化法同样是实现二芳基乙烯光开关分子可见光驱动的有效方法。早期研究表明,将过渡金属(包括Re、Pt、Ru 和Ir)与二芳基乙烯进行络合,利用金属-配体电荷转移(MLCT)激发态可以成功敏化二芳基乙烯分子。近年来,有机小分子敏化剂(例如黄酮和1,4-丁二酮)也被证实可以用来敏化二芳基乙烯分子。然而,由于n→π*跃迁强度较低(对称禁阻),已报道的有机三线态敏化效率通常较低,且往往需要添加过量的三线态敏化剂。为了克服这一弊端,Hecht等[50]巧妙地将1,4-丁二酮通过烯键偶联到二芳基乙烯侧链噻吩环,如图8(a)所示。得益于侧链共轭延伸,DAE-33o的最大吸收波长位于405nm附近,且呈现出极强的吸收带,因此闭环过程可以通过可见光进行驱动。闭环反应效率具有氧气依赖特性且能够被蒽(较低三线态能级)有效淬灭,证明该体系存在三线态敏化过程。随后作者借助纳秒/微秒瞬态吸收技术,进一步证明了三线态敏化过程在该变色体系中的主导作用。此外,虽然目标化合物的开环量子产率降低至ΦC→O=3×10-4,但通过三线态途径闭环可以有效避免副产物的生成,显著提升光致变色抗疲劳性。
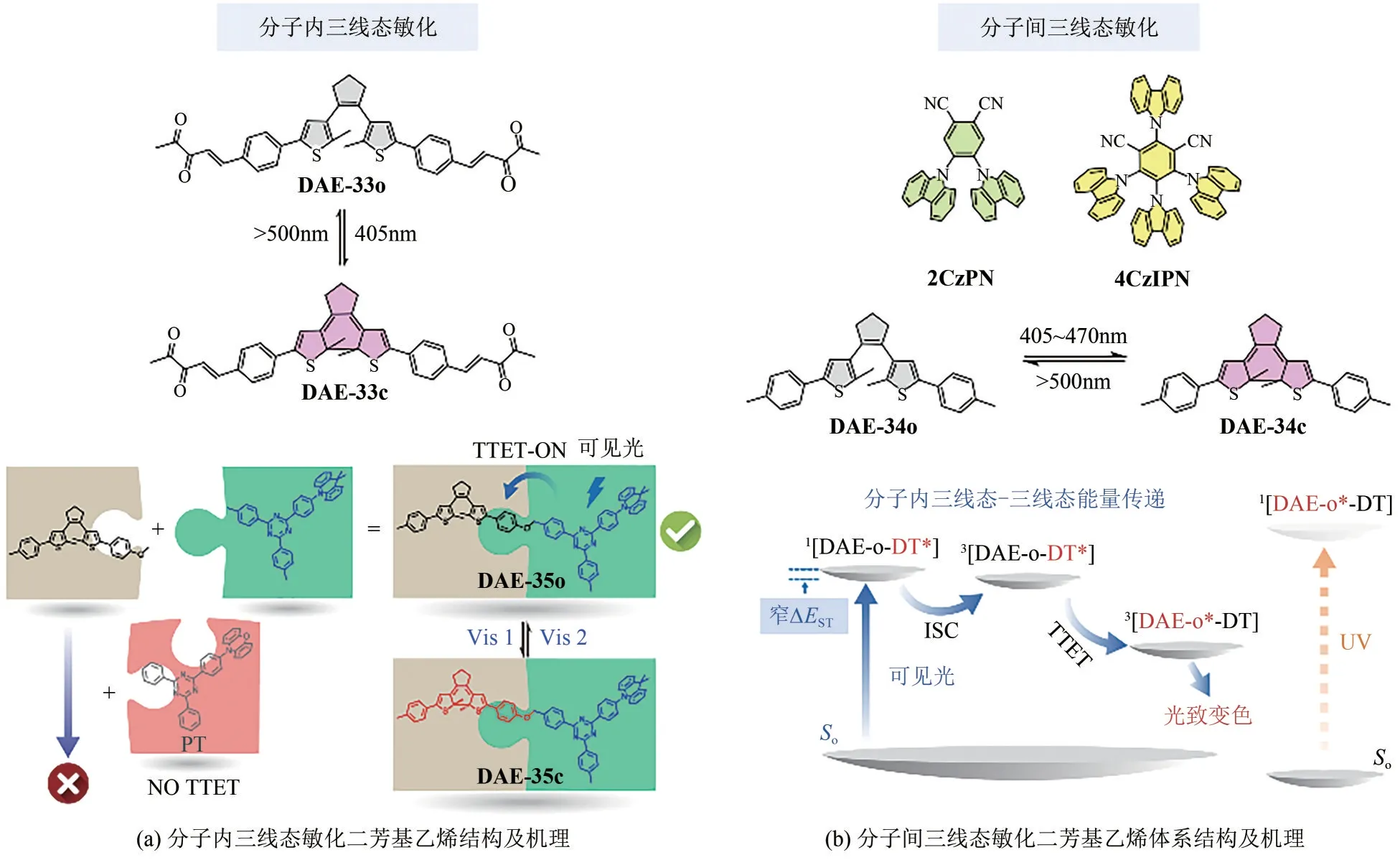
图8 三线态敏化可见光二芳基乙烯分子结构及原理示意图[50-52]
如此前所述,传统的全有机三线态敏化剂在可见光区吸收通常较低(甚至没有),导致可见光光致变色效率低下。用于敏化二芳基乙烯的完美有机敏化剂应满足以下条件:①敏化剂的单线态/三线态(S1/T1)能级应位于二芳基乙烯单/三线态能级之间,确保三线态敏化过程顺利发生;②敏化剂单线态激发所需能量应降低至接近三线态能量,确保激发波长最大程度红移;③敏化剂应具有高的系间窜跃(ISC)效率和高摩尔消光系数。为了实现这一目标,本文作者课题组[51]创新性地采用具有窄单-三线态能隙(ΔEST)的分子作为敏化剂,首次报道了可见光驱动的纯有机二芳基乙烯光开关体系,如图8(b)所示。通过将传统二芳基乙烯分子与敏化剂2CzPN或4CzIPN(2~3当量)简单混合,借助于三线态能量传递过程,DAE-34可以发生双向高效可见光光致变色。通过更换所选敏化剂分子,成功实现了二芳基乙烯分子在不同激发波长(405~470nm)下的高效驱动。此外,本文作者课题组还成功将该策略应用到聚合物薄膜中的“擦-写”应用探究。该方法简单方便、灵活高效,避免了传统方法复杂的分子设计与合成,为可见光光致变色提供了一种新策略。
不久之后,本文作者课题组[52]又进一步展示了基于三线态敏化机制的可见光二芳基乙烯模块化设计策略[图8(a),底部]。将具有窄单-三线态能级差的敏化剂DT(ΔEST=0.01eV)与传统二芳基乙烯分子共价相连,成功构建二分体DAE-35。共轭非共价的设计确保了敏化剂和二芳基乙烯部分的电子独立性,阻断了分子内可能的电子相互作用。在可见光激发下(λ=420nm),DAE-35 表现出高效的光致变色(ΦO→C高达0.40)和良好的抗疲劳性。此外,分子内三线态-三线态能量传递机制通过瞬态吸收光谱测试得到证明。通过更换能级匹配的敏化剂与二芳基乙烯分子可以实现光开关分子的模块化设计,有望用于构建光控电子材料及生物系统。
1.3.3 分子内质子转移法
分子内质子转移(intramolecular proton transfer,IPT)是一个普遍存在于氢键体系中的现象,如在水杨基亚胺类化合物中,活泼氢在质子给体酚羟基与质子受体亚胺键之间存在动态转移平衡过程。当活泼氢从氧原子转移到氮原子上,即从OH-构型转移到NH-构型时,化合物吸收波长会发生红移。如图9(a)所示,Zhu 等[53]通过在二芳基乙烯侧链部分引入IPT单元,成功构建了一系列可见光驱动的二芳基乙烯分子(DAE-36~DAE-39)。由于存在IPT 过程,该类分子开环体吸收带出现显著红移(400~500nm),使得可见光激发成为可能。以DAE-36为例,在450nm的可见光照射下,化合物发生快速的光闭环反应,闭环转化率高达95.6%,闭环量子产达到0.319,如图9(b)所示。尽管化合物在可见光下的闭环量子产率略有下降(Φ365nm=0.465),但抗疲劳性显著增强,即使连续照射2000min 后,仍未监测到闭环体的明显衰减[图9(c)]。密度泛函理论(DFT)计算结果显示,IPT 过程诱导的OH-构型到NH-构型转变有效降低了光环化反应的能隙(0.81eV),从而实现可见光激发。此外,该类新型全可见光二芳基乙烯分子在聚合物凝胶介质中同样可以发生高效光致变色,展现出广阔的应用前景。

图9 基于IPT过程的可见光二芳基乙烯分子结构式及其光开关性能汇总[53]
1.3.4 上转换/多光子吸收法
近红外驱动的二芳基乙烯体系可以通过上转换过程或多光子吸收间接实现。Branda等[54]借助上转换纳米粒子通过原位调节980nm激发光的强度实现了传统二芳基乙烯的高效开闭环反应。Irie 等[55]将具有多光子吸收特性的三苯胺单元与二芳基乙烯共价相连,通过使用单一的红外光(1.28μm)成功对二芳基乙烯的开闭环过程进行驱动。
2 新兴光开关染料
2.1 靛蓝
靛蓝类光开关分子包括以靛蓝染料为母核的一系列衍生物。靛蓝染料母体存在顺式、反式构型,且二者的吸收均在可见光区。除了可见光响应特性外,靛蓝光开关分子还具有量子产率高、光稳定性强、抗疲劳性佳等优点。近年来靛蓝类衍生物在构建长波长光开关分子领域备受关注,主要分为母体靛蓝、半靛蓝(HI)和半硫靛蓝(HTI)三类衍生物。
2.1.1 推-拉体系
传统的靛蓝染料分子由于存在激发态质子转移(ESPT)过程,光致变色受限[56]。通过引入烷基或乙酰基可以完全消除ESPT 过程,染料分子可以在可见光照射下发生顺反异构化,但是所得亚稳态顺式异构体的热稳定性极差,半衰期通常低于1s。最近,Hecht等[57]报道了一系列红光驱动的N-烷基和N-芳基取代靛蓝染料,通过π 共轭延伸及其推拉电子效应,成功实现了对顺式异构体半衰期的高效调控(从秒到小时)。如图10所示,通过引入具有吸电子性能的酯基,化合物40 反式和顺式异构体吸收带明显分离,二者最大吸收波长差值(Δλ)达到62nm,同时热稳定性显著增强,半衰期达到2.8min。将取代基从烷基变为芳基,染料分子E-型异构体的最大吸收波长几乎不受影响,这是由于芳基单元与靛蓝母体平面呈垂直正交构型造成。值得注意的是,由于Z型异构体中芳基间存在π-π相互作用,而E型异构体芳基与邻近的羰基间存在斥力作用,因此通过微调芳基结构,可以有效调节光开关分子Z型异构体的热稳定性,例如,苯甲氧基取代的化合物41半衰期为58s,而化合物42(硝基苯取代)的半衰期长达408min。
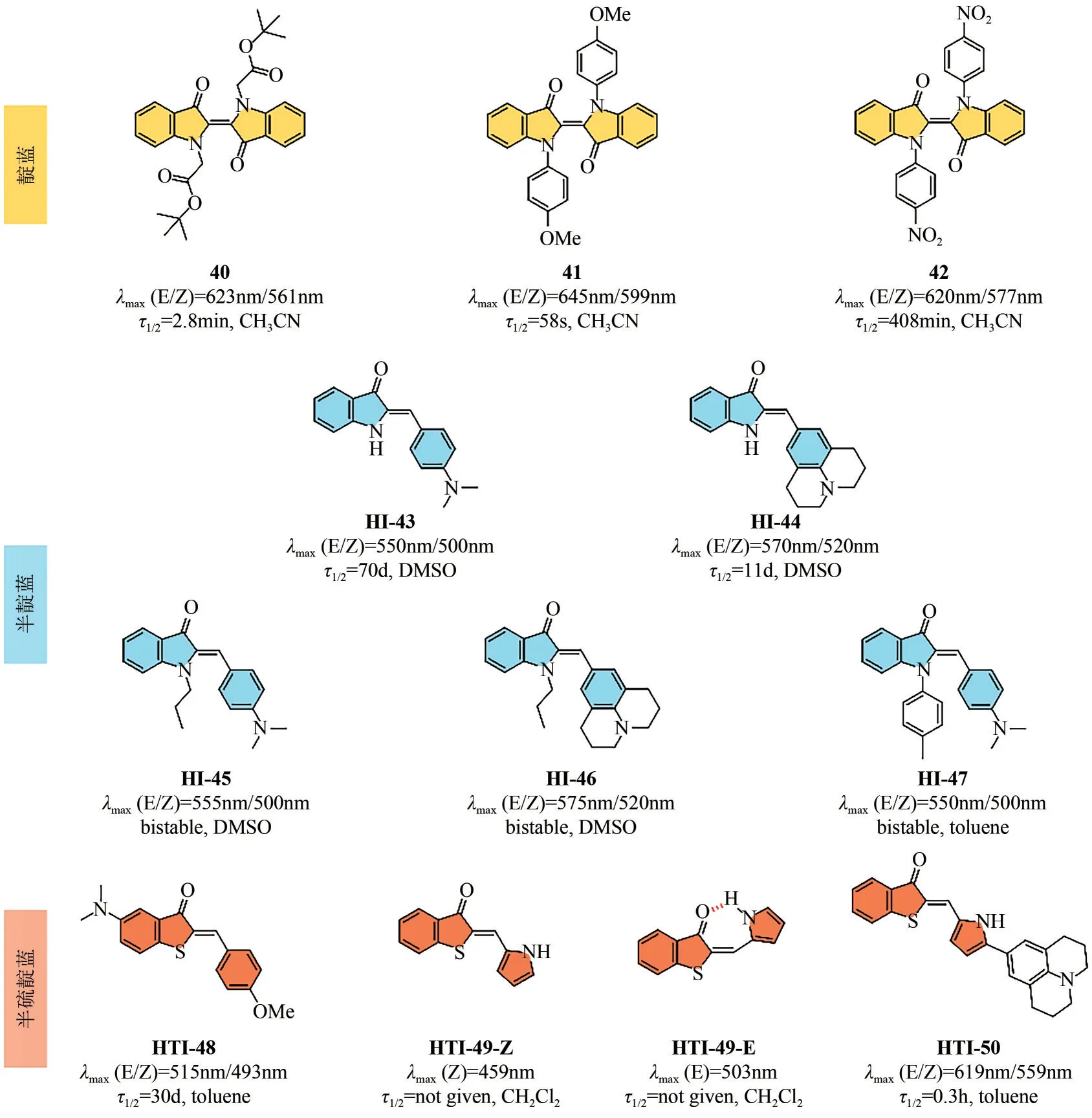
图10 可见光驱动靛蓝类光开关分子结构及其性能汇总
半靛蓝染料是由靛蓝染料与二苯乙烯片段组成,二者共享中心双键,并表现出与母体靛蓝相似的特性。自从半靛蓝染料的光异构化现象被首次报道以来,如何提升其光致变色性能成为研究热点。Dube 等[58]在二苯乙烯苯环片段上引入强供电子取代基,成功构建了基于“推-拉”电子体系的长波长驱动半靛蓝光开关分子(如图10所示)。化合物HI-43~HI-47的Z型异构体吸收均超过500nm,同时E 型异构体最大吸收波长位于黄绿光区域(>550nm)。分别使用蓝光/绿光、黄光/红光可以激发该类分子Z→E、E→Z 异构化过程,转化率分别>90%、>95%。其中化合物HI-46 的光开关性能尤其突出:在470~505nm的光照射下,E型异构体的产率大于95%,而在红光(680nm)光照下,所得E 型异构体几乎完全转化为初始Z 型异构体(图11)。此外,上述化合物均呈现出较高的热力学能垒。例如,HI-43 和HI-44 的E→Z 过程热力学能垒高达22.2~25.5kcal/mol(1kcal=4.18kJ),光照后所得E型异构体在室温条件下足够稳定,属于典型的双稳态光开关。通过进一步对羟基吲哚单元N原子进行修饰,化合物HI-45~HI-47 的热稳定性进一步提升,其中HI-46的半衰期长达83年。
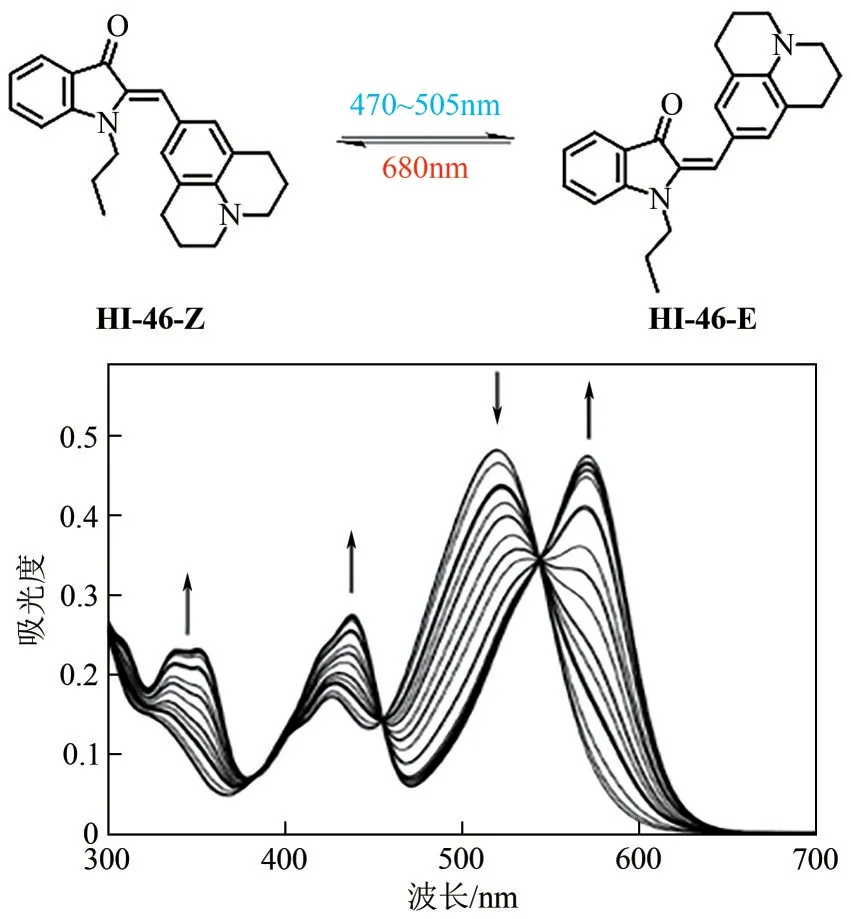
图11 HI-46的异构化过程及对应的吸收谱图变化[60]
半硫靛蓝染料是另一种常见的靛蓝衍生物,是由硫靛蓝结构与二苯乙烯结构共享碳碳双键融合形成[59]。为构建红光驱动的半硫靛蓝光开关分子,Dube 等[60]通过在二苯乙烯苯环片段上引入强给电子基团,加强中心双键的供体-受体特性,实现了吸收波长的显著红移。然而,该策略同时导致其E型异构体热稳定性大幅下降。通过对硫靛蓝基团进行适当修饰,可以有效实现激发波长及热稳定性的协同优化。化合物HTI-48的光异构化过程可以分别用绿光(505nm)和红光(高达625nm)高效激发,同时其E 型异构体的半衰期达到30 天(25℃),对应的热力学驰豫能垒为26.5kcal/mol(1kcal=4.18kJ)。
2.1.2 分子内氢键作用
Newhouse等[61]利用分子内氢键作用构建了长波长驱动的双稳态半硫靛蓝光开关染料分子。与苯环及杂芳环取代相比,吡咯取代的HTI-49分子由于存在分子内氢键作用,HOMO-LUMO 能隙降低,E 型异构体最大吸收波长红移至503nm(图10)。分子内氢键作用同时诱导光开关分子E/Z型异构体π-π*吸收带显著分离,提升光开关性能。因此,HTI-49 的Z→E 及E→Z 的异构化过程可以分别用蓝光(459nm)和绿光(503nm)触发,且转化率均接近于100%。在此基础上,通过在吡咯基团5号位扩展分子π共轭体系,可以进一步红移光开关激发波长。构效关系研究表明,HTI-49 的E/Z 型异构体在系列化合物中均展现出最长的吸收带。尽管转化率略有降低(62%),HTI-50异构化反应仍可以被黄绿光(567nm)和近红外光(740nm)分别激发。
2.2 二氢芘
二氢芘(DHP)是另一类新兴的基于己三烯环化的可见光光开关染料分子。由于相对较差的热稳定性及复杂的合成过程,二氢芘此前并未受到广泛研究。DHP 在可见光光照下发生典型的负光致变色现象,即相对于不稳定的形式,热力学稳定的异构体激发能量更低,是一类潜在的长波长光开关母体分子。近年来,基于DHP 的新型红光/近红外光致变色染料分子屡有报道[62]。通过向二氢芘母体结构引入推拉电子基团,可以有效红移分子激发波长至近红外区。
Hecht 等[63]报道了一种基于二甲基氨基/硝基的“推-拉”型叔丁基-二氢芘分子DHP-51(图12)。将电子供/受体基团与芘单元共轭连接,得到的芳香共轭结构同时存在醌类共振结构(ca.10%),吸收带因此显著红移(>800nm)。在近红外光(780nm)照射下,DHP-51在乙腈溶剂中表现出负向光致变色:即最大吸收峰逐渐下降,同时在较短波长(290nm)处形成新的吸收带,该光反应接近定量转化(>95%),量子产率为0.05。值得注意的是,DHP-51 的光开关性能表现出很强的溶剂化效应。在大极性溶剂中,例如DMSO/水的混合物(8∶2),闭环异构体DHP-51 的最大吸收波长进一步红移,其尾部延伸至835nm,开环量子产率约为0.06,异构体半衰期为0.5h;在非极性溶剂(正庚烷)中最大吸收仅达到红光区域(720nm),但量子产率高达0.28,异构体半衰期达到23h。
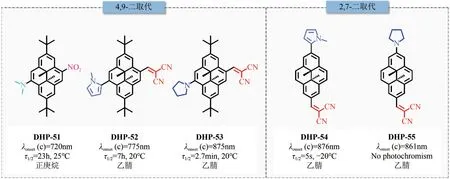
图12 红光/近红外二氢芘光开关分子结构及其性能汇总
在上述工作基础上,Hecht 等[64]对上述近红外光开关分子进行了构效关系研究,详细探究了取代基位置(2、7号位或4、9号位)对光开关分子性能的影响。所有衍生物DHP-52~DHP-55 在远红光或近红外区域(λ>750nm)均显示出极强的电荷转移吸收带。其中,DHP-52和DHP-54以N-甲基吡咯作为电子供体,丙二腈单元作为电子受体,二者的吸收带尾分别延伸至900nm 和800nm。DHP-54中由于存在较大的空间位阻,N-甲基吡咯单元扭曲至芳香平面外,共轭程度相对降低,吸收带较DHP-52 发生蓝移,表明取代基位置对CT 吸收带有显著影响,如图13 所示。化合物DHP-54、DHP-52 分别可以在780nm、730nm LED 灯照射下发生负向光致变色,转化率分别为10%、>97%。转化率的差别与二者异构体的半衰期长短相一致,DHP-54 开环体半衰期仅为5s(-20℃),而DHP-52 开环体半衰期长达7h(20℃)。DHPs 的光致变色特性还可以通过更换供体、受体单元来调节。当引入给电子能力更强的吡咯烷基时,DHP-53和55的整体吸收谱图发生明显红移。其中,DHP-53的吸收带红移至900nm,可以在810nm的LED光诱导下发生开环过程,开环转化率>94%,然而异构体半衰期从7h 降低至2.7min。计算结果表明,活性碳间的碳-碳单键在开环过程中起着关键作用,在醌类共振式中,碳-碳键相对较弱,导致光异构化效率较低。需要注意的是,尽管通过引入更强吸电子基团可以进一步红移化合物吸收波长至>1000nm,但该种情况下化合物光致变色性能会受到损害甚至被完全抑制。因此,采用“推-拉”策略构建近红外二氢芘激发波长存在上限,需要对多种影响因素进行综合考虑。
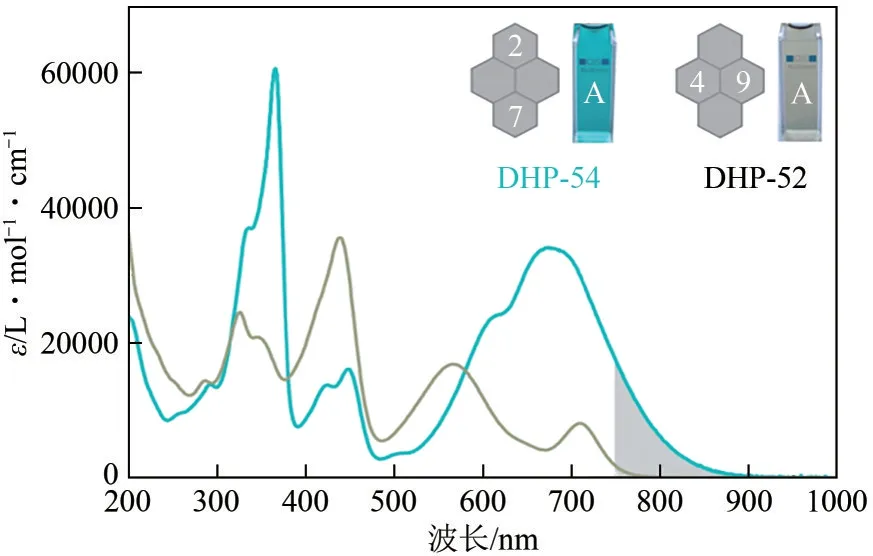
图13 化合物DHP-52,54溶液颜色及对应的吸收谱图[64]
2.3 给体-受体Stenhouse加合物
给体-受体Stenhouse 加合物(DASA)是近年来出现的一类新型可见光光开关染料分子。该类光开关分子制备简单且具有独特的负向光致变色性能,近年来引起了广泛关注[65]。DASA 可以在可见光照射下经过Z-E 异构化和随后的4π 电环化,由初始的有色态转化为致密的无色状态(图14)。到目前为止,已有三代DASA被开发报道。

图14 三代DASA分子结构式及其光开关性能汇总[65]
Alaniz 等[66]于2014 年首次报道了DASA 类光开关分子。第一代DASA 分子以不同的次级脂肪族、环状或者功能性胺基作为供体基团,麦氏酸(Meldrum’s acid)或巴比妥酸(Barbituric acid)作为相应的受体单元。由于强的CT特性和共轭延伸,DASA 在可见光区域(400~650nm)显示出强烈的吸收带[66]。构效关系研究表明,烷基胺种类对DASA 分子最大吸收波长几乎无影响,而受体基团对分子吸收波长影响显著。当受体由麦氏酸(DASA-56)改变为1,3-二甲基巴比妥酸(DASA-57)后,光开关分子的最大吸收波长由545nm红移至570nm。在可见光(545nm 或570nm)照射下,DASA-56、DASA-57 均可以由初始的有色态完全转化为无色的闭环异构体,且具有良好的抗疲劳性(图15)。停止光照后,所得闭环体在芳香族溶剂中快速热弛豫恢复至初始状态,表明DASA 属于T型光开关分子。DASA-56 经10min 即可恢复至初始态,而DASA-56则需要20min,说明DASA的热弛豫速率同样受到受体基团的显著影响。第一代DASA 分子美中不足的是激发波长均在600nm 以下,且光开关性能依赖甲苯等非极性溶剂,在极性溶剂或固体基质中性能受限。
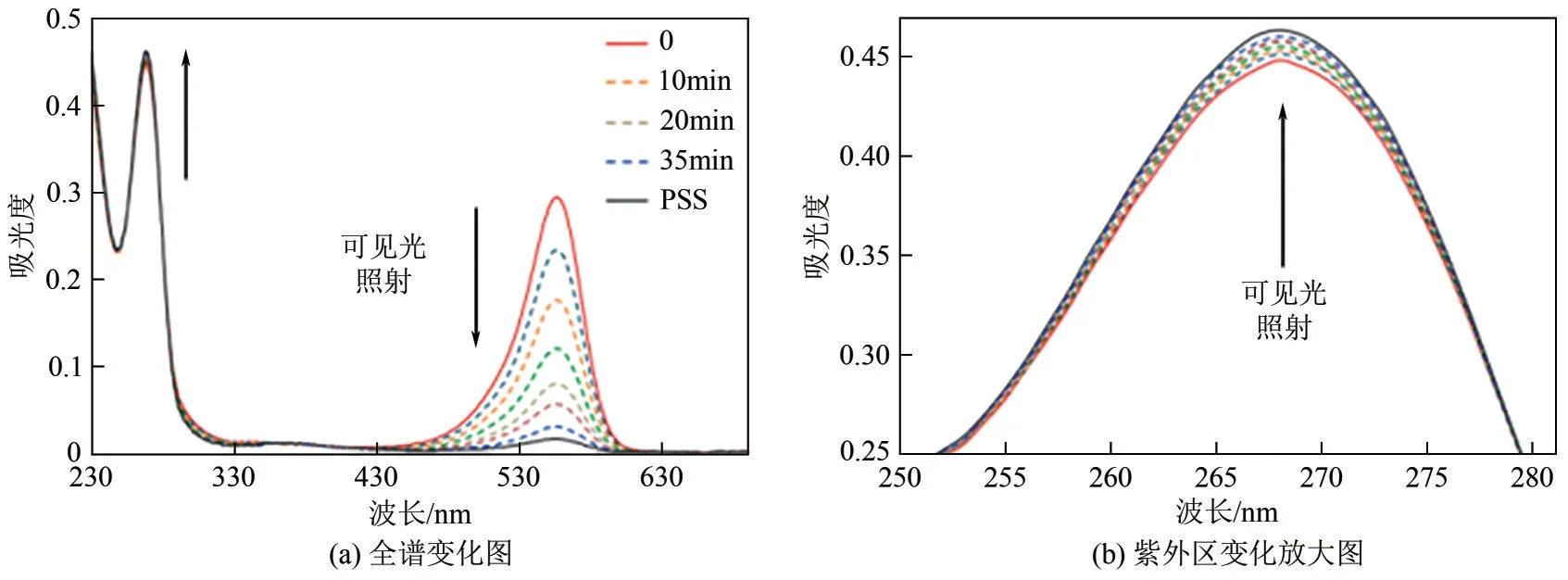
图15 DASA-57在可见光照射下的吸收谱图变化[66]
Alaniz 等[67]随后报道了第二代DASA 光开关分子,即采用二级苯胺衍生物作为供体基团,增强分子内电子推-拉作用。烷基胺被N-甲基苯胺、四氢喹啉和吲哚基供体取代,所得DASA最大吸收波长分别红移至582nm、599nm 和615nm。此外,相比于原始吲哚DASA(λmax=615nm),通过在环状苯胺的对位引入给电子基团,可以进一步红移分子最大吸收波长。例如,当引入甲氧基和N,N-烷基后,所得DASA-58、DASA-59的最大吸收波长分别红移至629nm 和669nm。在环状苯胺DASA 中,供体-受体之间分子轨道杂化过程可能对分子共轭程度有增强作用,从而导致吸收红移;而在非环状N-烷基取代苯胺类DASA中,对位取代基团对分子吸收波长几乎无影响(约2nm),造成该现象的原因可能是供体和受体之间较大的二面角(ΦD-A≈40°)阻止了电荷转移过程。与第一代DASA不同,第二代DASA 在极性溶剂(如THF、DCM、乙腈)中展现出良好的光敏性,在近红外光的驱动下可以发生当量光转换,同时呈现出优异的抗疲劳性。然而,该类分子在黑暗条件下的热力学平衡发生显著移动,有色的开环体异构体比例显著降低(2%~70%)。
为了调控黑暗条件下异构体平衡比例,进一步优化光开关特性,Alaniz 等[68]采用了不同种类的酸结构作为受体单元,设计并合成了第三代DASA。类似地,该类DASA较第一代吸收波长发生明显红移(585~680nm)。DASA-60 以羟基吡啶酮作为受体,最大吸收波长红移至680nm,在CDCl3中吸收尾部延伸至720nm,因此在近红外光照射下可以发生高效的光异构化,并且在甲苯、辛烷体积比为3∶1 的混合溶剂中同样表现出优异的抗疲劳性。更为重要的是,黑暗条件下第三代DASA开环异构体所占比例大都>75%,其中DASA-61中开环体所占比例高于95%。三代DASA体系为设计可见光/近红外光开关功能分子提供了基础,在药物传递、液晶、光图案和化学传感等方面具有潜在应用价值[69]。
3 结语与展望
过去十年中,各种新型光开关染料分子层出不穷,在光响应致动器、场效应晶体管、智能柔性材料等领域展现出了巨大应用前景[70-74]。其中,长波长驱动的光开关分子由于避免了高毒性紫外光的使用,满足光药理学等化学生物领域应用需求,受到广泛关注[17-19]。然而,这类光开关分子大多数激发波长仍位于400~600nm 窗口内,较低的组织穿透深度(<1mm)致使相关生物应用仍然局限于体外研究或浅表器官(如皮肤)。因此,实现光开关分子双向红光/近红外(650~1450nm)驱动是推进其活体应用的关键所在。现阶段已报道的红光/近红外光开关大多基于负向光致变色体系,该类分子可以在“生物光学窗口”内发生异构化,但逆向异构化只能依赖于热弛豫过程,且亚稳态异构体的热半衰期(τ1/2)往往会随激发波长的红移而下降[63]。虽然较短半衰期在光药理学的特定场景下可以减少光控药物副作用[75],但无法最大限度发挥“生物光学窗口”下高时空分辨操作的优势。因此,开发双稳态红光/近红外光开关染料分子显得尤为重要。
二芳基乙烯是一类典型的双稳态光开关分子,通过π-共轭延伸策略可以有效红移吸收带。然而,采用π-共轭延伸或给体-受体策略实现吸收红移,通常会不可避免地消除或显著降低LUMO轨道对激发跃迁的贡献,从而对光异构化过程产生不利影响。迄今为止,长波长二芳基乙烯分子设计仍然缺乏高效通用的设计策略,致使其研究相对滞后。随着计算化学和机器学习的快速发展,科学家们有望建立更准确的模型以精准预测分子结构及其光化学行为,革新光开关分子设计过程。
三线态敏化可以通过低激发态三线态过程激活光开关。借助于三线态敏化剂与光开关分子间的能量传递,可以有效避免光开关分子复杂的结构修饰,从而实现目标分子的长波长激发,维持原有分子的优良光开关性能。然而,三线态敏化策略容易受体系氧气浓度及分子间距离(由于Dexter电子交换机制)影响。将三线态敏化光开关系统集成到封闭的环境中,如密集排列的聚合物薄膜、胶束甚至纳米通道,有望克服上述弊端,推进三线态敏化体系在化学-生物-医学等交叉学科领域的具体应用。
最后,开发红光/近红外驱动的新型光开关母体分子同样尤为重要。DASA、靛蓝等自身吸收在可见光区新兴光开关分子的出现,显著推动了长波长光开关染料分子的研究进程。有理由相信,经过合理的设计与不断创新,更多新型长波长光开关染料母体会被成功开发,满足日益增长的活体应用需求。
猜你喜欢
系统仿真技术(2022年4期)2023-01-17
云南化工(2021年8期)2021-12-21
成都大学学报(自然科学版)(2021年1期)2021-05-22
国外医药(抗生素分册)(2016年4期)2016-07-12
信息记录材料(2016年4期)2016-03-11
中国粮油学报(2016年1期)2016-02-06
合成化学(2015年2期)2016-01-17
化工进展(2015年6期)2015-11-13
化工进展(2015年6期)2015-11-13
中国塑料(2015年10期)2015-10-14
