
电催化有机合成反应的活性和选择性调控研究进展
2023-10-07 12:34向阳黄寻魏子栋
化工进展 2023年8期
向阳,黄寻,魏子栋
(重庆大学化学化工学院,重庆 401331)
随着能源和环境问题的日益严峻,减少污染、发展可持续绿色化学显得尤为迫切。有机合成的产物在人们的衣、食、住、行中扮演着至关重要的角色,然而传统的有机合成往往伴随着高温高压或使用有毒试剂,与绿色发展的初衷背道而驰。近些年,有机电合成被视为一种绿色可持续替代传统有机合成方式的重要途径。该方法利用电作为一种清洁氧化还原剂取代有毒、危险的氧化还原试剂,实现电驱动的化学产品合成,具有反应条件温和、原子经济性高等特点,是目前合成化学的研究热点[1-2]。更重要的是,有机电合成中的电能可来自可再生能源发电,可实现可再生能源电力的消纳,成为“源网荷储一体化”的重要枢纽,助力“碳达峰”和“碳中和”的实现。
有机电合成可分为直接电合成和间接电合成[3-5]。间接有机合成利用电极表面产生的氧化还原电对作为反应媒介,反应媒介与底物进行反应后再在电极表面进行再生循环,避免了有机物与界面的直接接触,但存在电解效率低、媒介分子/离子回收困难等问题。直接有机电合成是指反应物在电极表面直接发生氧化还原,反应体系简单、高效,更适合大规模工业应用,是本文的主要研究对象。电催化有机合成反应主要发生在电极-溶液界面,反应物种在界面处的传质、吸附和反应以及产物的解吸决定了电化学反应的活性和选择性[6-7]。提高电催化活性,可以在相同原料处理量下降低反应器尺寸和催化剂用量,而提高选择性则有助于降低副产物生成和分离能耗,两者共同决定了电催化反应过程的效率和经济性。根据尺度的不同,电催化合成的活性和选择性调控策略主要包括以下几种:① 调控催化剂电子结构,促进反应物的定向吸附与活化;② 调节电极-溶液界面组成,选择性富集反应物分子/离子;③ 强化反应与传递过程,加快反应物种的结合或分离。本文从以上几个方面,综述了近年文献中所报道的选择性调控方法,并讨论了存在的问题和未来的研究思路。
1 催化剂电子结构调控
与电解水、氯碱工业等无机过程相比,电催化有机合成活性与选择性不但与反应物分子的吸附强度有关,而且与其吸附构型相关,并且通常涉及几种反应物或中间体的共吸附过程。催化剂的电子结构直接影响反应物种在活性位点的吸附形式,进而影响反应活性和选择性。目前,关于电催化剂电子结构调控方法已有较多研究,包括合金化、掺杂、异质结等策略。本节主要介绍上述方法在提升电合成活性和选择性方面的应用。
1.1 合金化
合金化通过具有不同电负性金属元素间的电子转移,调节活性位点的电子结构,使之与反应物种的吸脱附更匹配[8]。例如,施剑林院士团队[9]采用溶剂热法合成了PdAg/NF合金催化剂用于乙二醇氧化。其中,Pd 作为乙二醇电氧化活性位点,而Ag的加入调整了Pd 的d 带中心,使乙醇酸易于从Pd表面解吸,防止过度氧化,从而提高选择性。与传统合金化相比,高熵合金(HEAs)除了具有优异的物理和化学性质(抗氧化、耐腐蚀、高强度、耐磨性等)外,多元素随机分布在单一HEA 相中,使其发生晶格畸变和电子结构改变,被广泛用于储能和电催化材料[10-11]。Fan等[12]通过原位生长结合热还原制备出HEA-CoNiCuMnMo 纳米颗粒自支撑电极。与四元合金相比,HEA-CoNiCuMnMo 催化剂在甘油中电催化活性明显增强。机器学习模拟揭露相边界富集的Mo、Ni 和Mn 改变了合金的电子结构,得到了最佳吸附能,从而获得了优异的催化性能,在宽电位范围(1.27~1.47Vvs.RHE)内,甲酸酯产物具有超过90%高选择性。虽然合金化能够调节催化剂的电子结构和形貌特征,但精确控制合金化的比例和结构(尤其是高熵合金)依然面临巨大挑战。
1.2 构筑异质结
通过形成异质结,可以产生内建电场和两个相反的电荷分布区,内建电场可以促进催化过程中电子传输提高催化活性,而特殊的空间电荷区影响着反应物/产物的吸附和脱附,可调节目标产物的生成[13-14]。例如,Yang 等[15]以原位生长结合低温磷化的方法合成了具有异质结构的MoO2-FeP 作为析氢和5-羟甲基糠醛(HMF)氧化的双效催化剂,电子从MoO2转移至FeP,从而电子富集于FeP相,使其具有优异的H2O 和H*吸附能,而缺电子的MoO2更有利于HMF的氧化,在转化率100%时产物2,5-呋喃羧酸的选择性达到98.6%。Yuan 等[16]利用Mott-Schottky异质结特殊的空间电荷区,针对性地制备了Bi-BiVO4异质结催化剂用于合成尿素。该催化剂界面电荷分布形成局部亲电亲核区,促使惰性分子CO2和N2分子的靶向吸附和活化,抑制了CO 中毒和*NNH 中间体的形成,保证了*N= = N*中间体的偶联,生成尿素分子前体*NCON*中间体。在0.1mol/L KHCO3(-0.4Vvs.RHE)条件下,尿素产物高达5.91mmol/(h·g),法拉第效率为12.55%。
1.3 掺杂改性
非金属改性能够有效地调控活性中心金属元素的电子密度,从而促进反应过程中电子转移,提高反应活性和产物选择性。Huang 等[17]采用热分解法制备了RuO2-SnO2-TiO2/Ti 电极用于肉桂醛选择性加氢制备肉桂醇,在58%转化率下的选择性达到了88.86%,远高于单金属电极(<15%),其原因在于Ru的部分电子转移至O,形成局部Lewis酸中心,选择性吸附极性C= = O 键,抑制了热力学更容易发生的C= = C 键加氢(图1)。Sheraz 等[18]采用还原和水热相结合的方法制备了N、P共掺杂的Pd碳纳米管催化剂用于甘油电催化氧化。由于N、P 的原子尺寸小,能够与Pd 形成间隙合金,更好地转移了电子,提升甘油电催化活性。此外,低非金属条件下(质量分数<25%),电荷转移从P原子转移至Pd原子,带负电荷的Pd可以有效地除去中间产物(CH3COO-)ads和(CO)ads,防止(CO)ads在催化剂表面积聚和C—C 键的断裂导致彻底氧化,促进甘油分子选择性转化为1,3-二羟基丙酮和中草酸。值得注意的是,对电催化氧化反应而言,掺杂的非金属元素容易被氧化电流氧化析出,这对催化剂的结构演变研究和催化剂稳定性提出了更高要求。韩布兴院士团队[19]设计的氮掺杂多孔碳负载的NiPt 和FeRu催化剂可实现同时高选择性地在阴极得到环己酮,阳极得到苯醌。在阴极,Pt位点富集了大量的电子云,阻止了羰基的吸附,避免了环己酮产物的进一步还原;在阳极,Fe修饰Ru改善了Ru表面的电场环境,使其具有更大的电子云密度,诱导电子从Fe 转移到Ru,阻碍了C= = C 双键在FeRu 表面上的吸附,防止苯醌产物发生过度氧化,因此得到较高苯醌选择性。此外,催化剂中氮掺杂多孔碳载体也为反应提供了优异的导电性和物质传输通道。
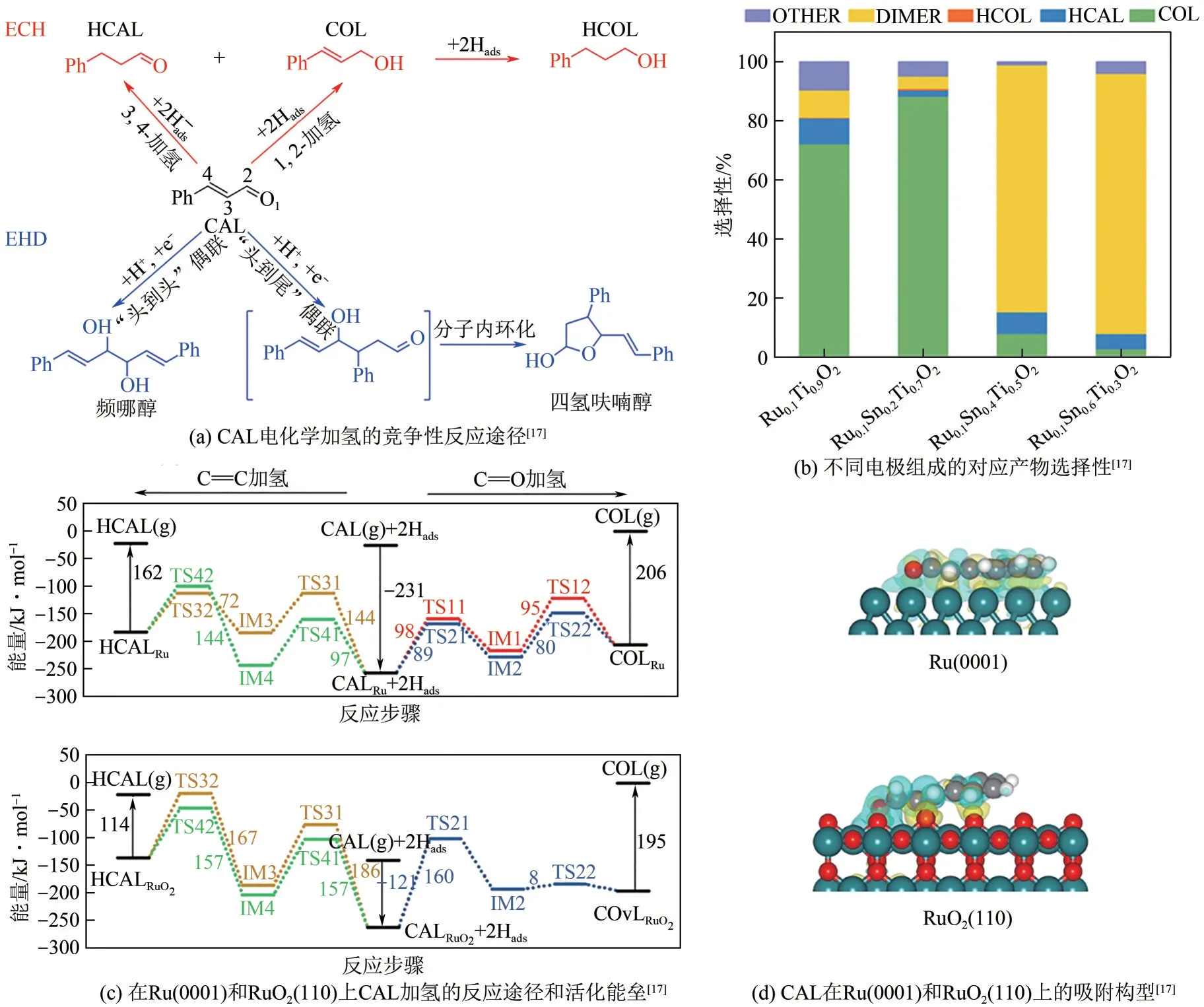
图1 RuO2-SnO2-TiO2/Ti电极用于肉桂醛(CAL)选择性加氢制备肉桂醇(COL)
同样地,金属掺杂可通过调节催化剂能带结构、表面物理和电子特性以及控制被吸附/脱附物质的结合强度,来增强电催化活性和选择性,在有机电合成中展现出广阔的应用前景[20-21]。例如,Sun等[22]采用水热合成系列金属(Mn、Co、Ir、Rh)掺杂的α-Ni(OH)2催化剂,用于苄胺电催化氧化。与其他金属不同,Mn的掺杂使OER过程的控速步骤(RDS)自由能变从0.87eV提升到1.30eV,从而降低了NiOOH 表面上析氧反应(OER)活性;改变了苄胺的吸附位点(Ni→Mn),降低了苄胺氧化RDS 的自由能变(图2)。苄胺分子和羟基在催化剂表面的共吸附平衡提升了苄胺选择性氧化,在转化率为99%时苯甲腈的法拉第效率高达96%。类似地,Zhou等[23]采用Mn掺杂CoOOH电催化剂,可以选择性地将木质素衍生物仲醇或酮氧化为羧酸。多金属催化剂可以通过协同效应改变反应物/中间体/产物在催化中心的吸附行为,从而提高某些反应的活性和选择性,使得在最佳反应条件下获得64.99%羧酸产率。

图2 在1.40V vs RHE和pH=13.5下,不同催化剂的自由能变图
1.4 引入氧空位
氧空位通过引入大量配位不饱和的缺陷位点,增强催化剂对某些有机分子中间体的特殊吸附,从而获得较高的目标产物选择性[24-25]。例如,Zhang课题组[26]报道了一种高效碳负载Ni/MoO2异质结催化剂,用于电催化苯酚加氢。引入MoO2后,Ni 与MoO2之间强电子相互作用增强了中间体的吸附,有利于反应的进行。苯酚在低氧空位的Ni/MoO2上能增强苯酚和中间产物的吸附,促进H的抽离加速了烯酮互变异构,生成动力学上有利的环己酮。氧空位浓度升高会促使环己酮中的氧占据MoO2中的氧空位,增强环己酮的吸附,使其进一步加氢生成环己醇[图3(a)、(b)]。因此,通过改变电催化剂中氧空位浓度可灵活调节产物选择性。Wei 等[27]报道一种富含氧空位的CeO2催化剂用于尿素合成。在该工作中,氧空位的引入能够锚定中间体*NO,促进其与*CO的偶联形成*OCNO,而抑制*NO的加氢反应,有助于尿素生成。该团队还将调节氧空位方法用于HMF 选择性氧化为2,5-呋喃羧酸[28],制备了富氧空位的Co3O4(VO-Co3O4)催化剂[图3(c)、(d)]。在该催化剂上,OH-和HMF在活性位上发生非竞争性吸附,即亲核OH-会被吸附到氧空位,并在氧化过程中与有机分子发生反应,降低脱氢控速步骤的反应能垒,提高尖晶石型氧化物的催化活性,降低反应过电位。氧空位除了调节中间体吸附外,还可以通过削弱键能来提升选择性。例如,Jia 等[29]合成富含氧空位的TiO2纳米管用于硝酸盐电还原合成铵。由于硝酸盐中的氧原子会填充TiO2中的氧空位削弱N—O键抑制副反应产物的形成,同时更多的氧空位可以调节反应中间体与催化剂之间的相互作用来提高铵的选择性。在最佳的电位下,法拉第效率和选择性分别达到85.0%和87.1%。虽然氧空位能提升目标反应活性和产物选择性,但在有机电催化中对氧空位的演化和再生研究较少。
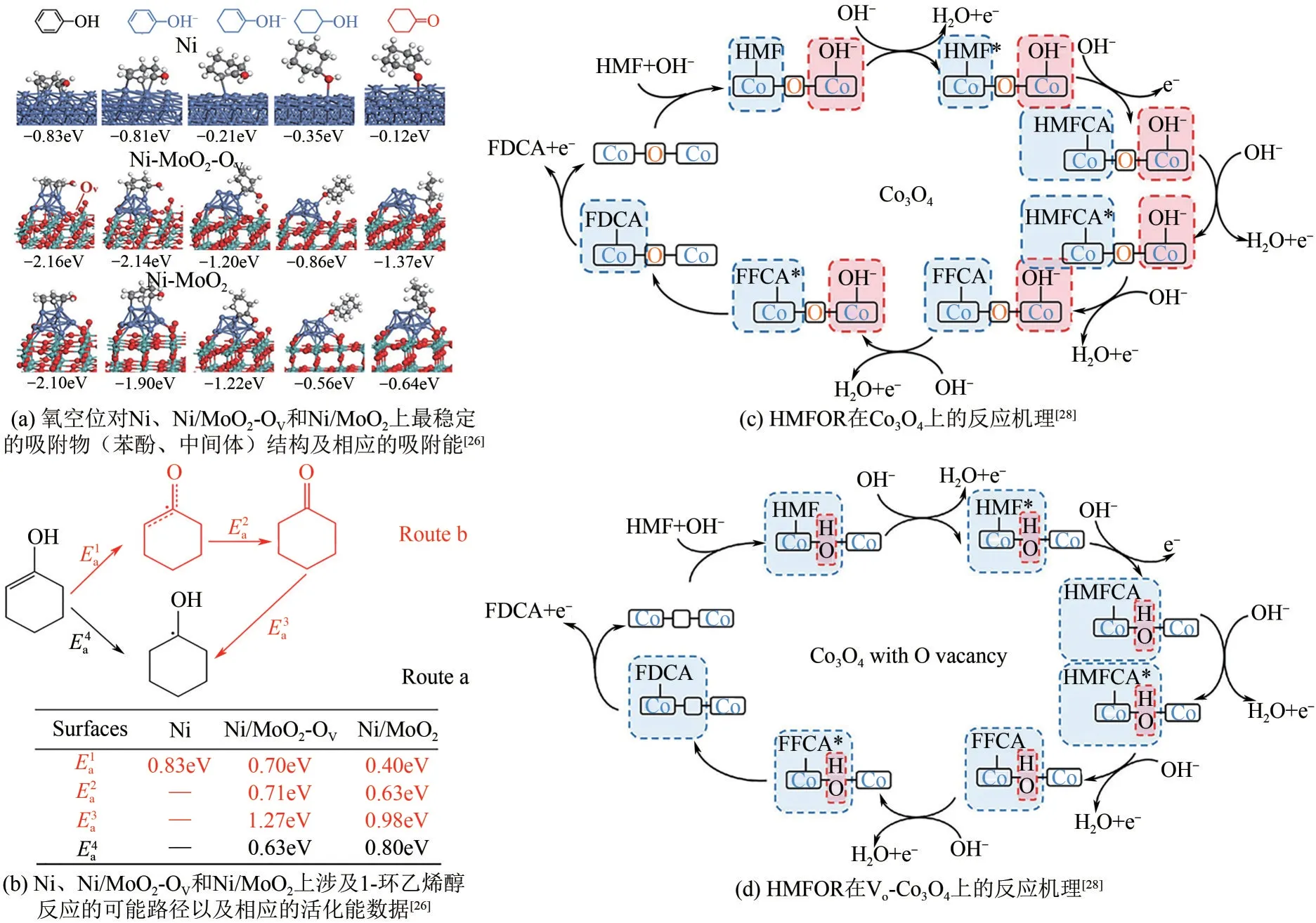
图3 相应的吸附能、反应路径及反应机理图
1.5 其他策略
针对有机分子的结构复杂性和特殊性[如复杂的手性分子、惰性的C(sp3)—H 键],除上述常规催化剂合成手段外,研究者还提出了一些特别的策略提升产物选择性。例如,催化剂的电子对称性也可对电化学反应选择性产生影响。Luo等[30]利用MnO2的电子自旋不对称结构,歧化苯乙烯成对π 电子,形成“双自由基”与活性氧结合生成苯甲醛,其选择性达到了65.02%(图4)。该作者进一步对比了在电子自旋稍不对称的MoO2催化剂,其苯甲醛选择性仅6.59%,而在电子自旋完全对称的MnO3和TiO2上,只生成苯甲酸。DFT计算表明,在自旋不对称的催化剂上吸附后,苯乙烯自身对称的电子结构也会被破坏,从而可通过Grob fragmentation 与OH、OOH等活性氧物种形成苯甲醛。
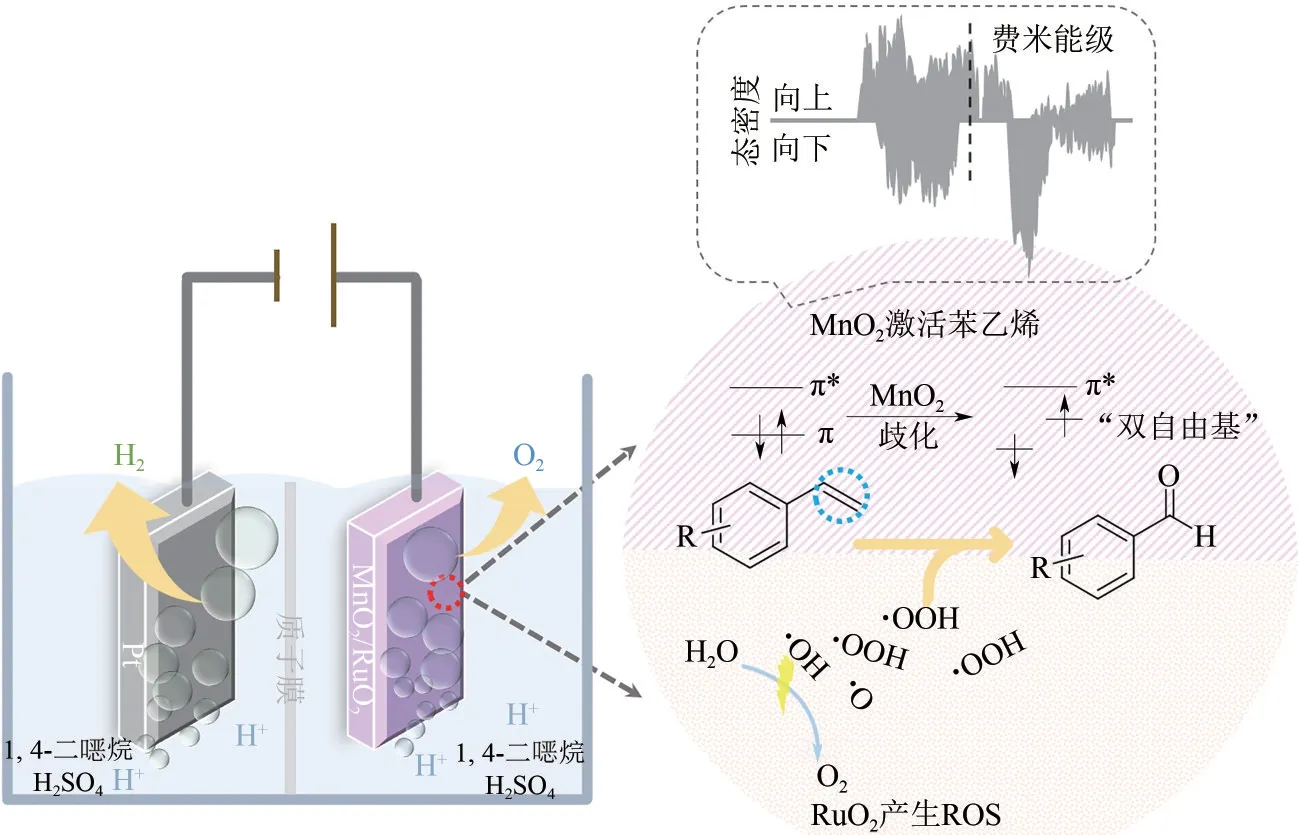
图4 MnO2/(Ru0.3Ti0.7)O2/Ti电极歧化苯乙烯成对π电子示意图[30]
对于部分阳极氧化反应,可通过催化剂某种元素中间价态的调节实现循环式间接氧化,从而达到高选择性的目的[31-32]。例如,Xin等[33]利用泡沫铜作为载体构建了钴氮共掺碳的纳米片阵列,该催化剂中的Cu+/Cu2+电对可以实现对葡萄糖氧化制葡萄酸的高选择性合成。与其他元素合成的催化剂(如Fe基催化剂[34]、CoNi合金[35]、CoNi过氧化物[36]等)相比,Cu2+具有特殊性,其起始电位远低于其他元素,这对指导合成电催化氧化葡萄糖的系列催化剂具有重要意义。Li等[37]采用共沉淀制备镍钒层状双金属氢氧化物纳米片(NV-LDH-NS)并将其用于苯酚电合成对苯二酚反应。NiV-LDH-NS内部可形成丰富的缺陷结构(VNi和VO)和低配位结构(V-O和Ni-O),加速了V4+/V5+氧化还原电对的循环,生成羟基自由基,促进苯酚的Csp2—H 键活化和羟基化。最终,该反应在72.2%转化率下可以实现71.6%的对苯二酚选择性。
2 电极-溶液界面调控
在电极催化剂活性组分不变的情况下,还可通过改变电极-油/水界面调控反应物在界面处的覆盖度和吸附构型,调节电化学反应活性和选择性,从而最大程度发挥活性位点的作用。以丙烯腈电化学还原反应为例,在工业反应条件下除生成二聚主产物己二腈外,还可能发生直接加氢反应生成丙腈,降低选择性和法拉第效率。Wu 等[38]揭示了丙烯腈在阴极反应途径与催化剂表面吸附组成的关系,即在没有活性氢的情况下,产物以己二腈为主;在存在活性氢的情况下,产物以丙腈甚至氢气为主。在实际反应过程中,即使采用高析氢过电位的Pb 或者Cd 也难以得到高己二腈选择性,因此可通过添加少量季铵盐在阴极表面形成疏水层,在抑制质子吸附的同时提高双电层内有机物浓度,从而获得较高的己二腈选择性[39-40]。
以水作为电催化合成溶剂可以大幅降低后续分离操作,更符合绿色化工的要求。然而,许多有机反应物在水中溶解性差,不利于电化学反应的进行。例如,在KA油(环己酮和环己醇混合物)氧化制己二酸中,原料环己酮仅微溶于水,限制了反应活性和产物的选择性。针对该问题,Li 等[41]在Ni(OH)2层间引入表面活性剂十二烷基苯磺酸钠(SDS),利用其表面疏水性使环己酮在催化剂层间发生富集,加快电化学反应的速度。结果表明,产物己二酸的生成速率提高了3.6 倍,法拉第效率从56%提升至93%(图5)。
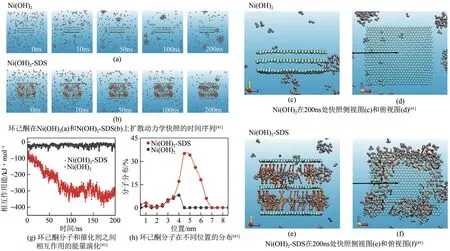
图5 环己酮在SDS修饰的层状Ni(OH)2上扩散行为的粗粒度分子动力学模拟
电催化氧化要求有机反应物和活性氧物种同时在电极表面发生吸附,但单一活性位点难以同时实现与有机物和活性氧的结合,设计具有两种活性位点的电催化界面是解决该问题的思路之一。Li等[42]制备了负载型Au/CoOOH用于苯甲醇氧化耦合电解水制氢反应,在1.5Vvs.RHE的低电位条件下实现了540mA/cm2的大电流密度,其原因在于Au 的5d空轨道与苯环π电子发生相互作用,实现苯甲醇的富集,同时CoOOH 产生大量活性氧物种,从而提高了苯甲醇的氧化活性。Wu 等[43]制备了包覆型TS-1@Co-N-C 催化剂,其中Co-N-C 壳是一种高效氧还原制双氧水催化剂,TS-1 分子筛则可以活化双氧水并与有机物的氧化反应。在氧还原耦合苯酚羟基化反应过程中,包覆型TS-1@Co-N-C 可以获得99.45%的邻/对苯二酚选择性,远高于机械混合的TS-1+Co-N-C 催化剂,其原因在于前者避免了H2O2在TS-1表面的降解。
在电极-溶液界面构建限域空间微环境,增强区域择形吸附性能,从而提升催化活性和产物选择性[44]。手性电有机化学是电催化和不对称合成的交叉学科,通过构筑手性电极、添加手性媒介(如手性溶剂、手性支持电解质等)、使用手性催化剂以及手性辅助等方式,实现手性产物的合成与分离[45-46]。例如,Yue 等[47]报道了一种用于催化芳香酮不对称还原的新型无金属手性电极,将具有手性的氨基酸通过酰胺键连接到多壁碳纳米管(MWCNTs)表面,发现L-精氨酸和D-精氨酸修饰后的阴极可分别获得(R)-和(S)-α-三氟甲基-苯甲醇,产率为60%~61%,对映体过量43%~44%,其性能可归因于手性修饰分子对产物形成的择形作用。
此外,溶液pH也会直接影响电极表面H或OH的覆盖度,进而影响产物分布。Hunsom 等[48]在探索甘油电催化氧化时发现,在不同的pH 下,产物的选择性也会发生变化:当pH 为1 和11 时,甘油脱氢生成甘油醛,然后C—C 键裂解生成乙二醇;而在强酸条件下丙酮和丙烯醛还原生成1,2-丙二醇和1,3-丙二醇。此外,甘油电氧化反应对中间产物和最终产物的选择性以及电催化剂(或电极)的活性也很大程度上取决于反应介质的pH。Verma等[49]比较了Au 电催化剂在碱性和酸性及中性中的甘油氧化性能。结果表明,在碱性条件下表现出较高的甘油催化活性,其主要产物为乙醇酸。而在酸性或中性条件下,Au电极表现出非常弱的电催化活性,只能生成少量的二羟基丙酮产物。Zhou等[50]讨论了糠醛在电催化氢化对pH 的依赖性机制。在酸性条件下,氧原子直接从氢化的糠醛衍生的烷氧基中间体中解离出来,然后通过热力学上有利的质子耦合电子转移过程逐步氢化形成H2O,从而获得了高比例的氢解产物(2-甲基呋喃);当H+不足的状态下,Oad很难通过Had转移生成H2O,从而阻碍了中性/碱性条件下2-甲基呋喃的生成,糠醛会直接氢化为糠醇(选择性约98%)。
3 反应与传递耦合调控
在有机电合成中,除了催化剂电子结构和电极-溶液界面组成外,传递问题是工业化过程中不可忽视的因素之一。其主要包括物质在电极表面的扩散、反应物和产物在电解质中的传质、电化学反应与传质过程之间的耦合关系等,它会直接影响到反应过程中反应物和产物的传输速率和浓度分布,从而对反应的选择性和法拉第效率造成影响[51-53]。通过合理设计电合成反应器结构和操作方式,协同强化反应、传递与分离过程,加快反应物种的传质或及时移除反应物,则可以进一步提高反应的效率和选择性,避免因传质缓慢导致的活性位点或反应界面利用率低等问题。通过多学科交叉,设计高效电合成反应器,是推动电合成广泛工业化的必经之路。
前文介绍了通过电极-溶液界面设计实现有机物在电极表面的富集,提升难溶性有机反应体系的效率。但在反应器尺度上,还需要实现反应物种的快速补充和产物的及时分离。Zhang 等[54]采用I-/I2作为氧化还原电对,并设计了一种连续流系统用于HMF 选择性电合成2,5-二甲酰基呋喃(DFF)。其中,阳极发生I-氧化生成I2,进入含有油(CH2Cl2)、水两相的反应池,I2被萃取至油相氧化有机底物,生成的I-进入水相并通过外管路返回至电解池阳极[图6(a)]。该自萃取方法促进了I-/I2向水/油相传质,而双相流系统将氧化剂的电化学再生与HMF 的氧化进行物理分离,避免了过度氧化,最终DFF 的选择性在99%以上。
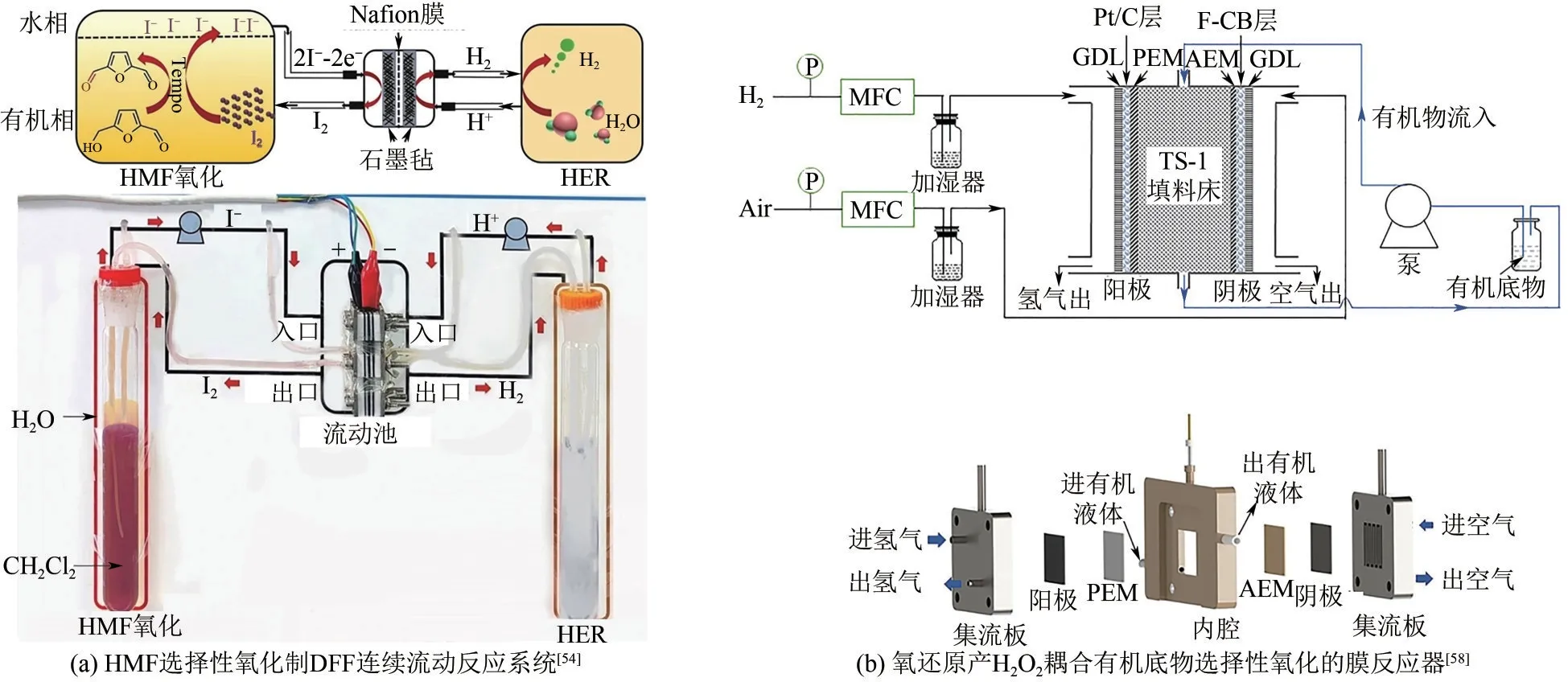
图6 电化学反应与传递耦合设计示例
膜分离是利用膜的选择性透过原理,实现不同组分的分离、纯化、浓缩的过程,具有高效、节能、过程简单等特征,可以很好地与电合成反应器进行结合[55-56]。Li 等[57]设计了一种电催化膜反应器用于正丙醇电催化,其阳极为兼具分离功能的负载纳米MnO2的微孔Ti隔膜。反应物从膜外向内渗透,丙酸从内部获得,可通过调节停留时间和反应温度提升正丙醇的转化率和丙酸选择性。
设计电合成反应器的特殊结构,实现电催化与热催化的联合,则可以充分利用成熟的热催化方法辅助提升电催化反应选择性。Zhang 等[58]设计了双膜微流电解反应器[图6(b)],用于氧还原产生H2O2和有机物选择性氧化。在碳材料作用下,阴极发生氧还原产生OOH-,阳极通过氢氧化或水氧化产生H+,两种离子分别通过阴、阳离子交换膜进入两膜之间的反应室生成H2O2,再在TS-1 分子筛作用下氧化有机物获得高附加值产品。该反应器应用于醇氧化、苯乙烯环氧化及环己酮氨肟化,在相同催化剂和反应器结构下都获得了较高的选择性和法拉第效率,表明该反应器对目标产物选择性提升具有普适性。由于过氧化物在阴极获得,因此阳极仍可用于底物直接电化学氧化,从而实现单一反应物(糠醛)和单一产物(糠酸)的新型成对电合成反应体系,总体法拉第效率高达132.79%。
电化学反应器中的电极间距不但影响溶液电阻,而且决定反应物和产物在电极间的扩散距离。Mo 等[59]设计了一种微流控的氧化还原中性电化学反应器,分别在两个电极产生不同的活性中间体。由于电极间距仅25μm,自由基在两极间的扩散可在亚秒时间内完成,远低于自由基的寿命,从而可在自由基分解前发生单电子自由基-自由基交叉偶联反应,获得高产物收率等。
4 结语与展望
利用电合成技术进行有机合成,既可以高效、高选择、绿色环保的合成目标产物,又可成为弃光/电的能源互补纽带,对我国节能减排具有重要意义。虽然研究者已经开发出多种提升有机电合成选择性策略,但仍面临着诸多挑战,包括有机电化学反应机理、催化剂与性能之间的构效关系不够明确,现有电合成反应器结构有待改进,反应与传递、分离的耦合需要进一步强化等。在今后的研究中,电合成领域需要通过多学科交叉和多尺度协同调控,打通微观反应机理、催化剂制备、电合成反应器设计以及工艺流程开发的微观至宏观研究路径,助力电合成反应的广泛工业化应用。
猜你喜欢
第一财经(2019年8期)2019-08-26
陶瓷学报(2019年5期)2019-01-12
中国有色金属学报(2018年2期)2018-03-26
中南大学学报(自然科学版)(2016年2期)2017-01-19
中国资源综合利用(2016年7期)2016-02-03
哈尔滨医药(2015年2期)2015-12-01
学习月刊(2015年14期)2015-07-09
物理化学学报(2015年5期)2015-02-28
读者欣赏(2014年6期)2014-07-03
语文知识(2014年2期)2014-02-28
