
基于UPLC指纹图谱和多成分定量的布渣叶饮片和标准汤剂量值传递研究
2023-09-25 07:18:48李振雨何民友刘晓霞曲丽媛周湘媛黄彩莹陈向东孙冬梅
天然产物研究与开发 2023年9期
李振雨,何民友,刘晓霞,曲丽媛,周湘媛,黄彩莹,陈向东,孙冬梅
1广东一方制药有限公司;2广东省中药配方颗粒企业重点实验室,佛山 528244
布渣叶为岭南特色药材,主要化学成分为黄酮、生物碱、萜类、有机酸等,具有促进胃肠蠕动和胃液分泌,降血脂,退黄,抗炎、镇痛、保护心血管,抗衰老等作用,为药食两用中药,最近几年对布渣叶的重视程度增加,研究也越来越多[1]。
传统中药以汤剂为主要用药形式,汤剂为中药发挥药效的物质基础,并经过上千年的临床实践,因此,建立中药标准汤剂的质量标准,能够为中药配方颗粒等现代中药制剂质量标准的建立提供重要参考[2]。目前,关于布渣叶标准汤剂的研究报道尚未见到,我司在进行布渣叶配方颗粒国家标准研究中,按照《中药配方颗粒质量控制与标准制定技术要求》(以下简称技术要求),对标准汤剂的关键质量指标:出膏率、浸出物、指标性成分含量及转移率、指纹图谱进行研究,考察布渣叶饮片到标准汤剂的量值传递规律,为布渣叶配方颗粒及其相关制剂质量标准的制定提供参考。
1 仪器、试剂与试药
1.1 仪器
Thermo超高效液相色谱仪(Vanquish,赛默飞世尔科技有限公司);Thermo Vanquish Flex超高效液相-Thermo Fisher QE高分辨质谱联用仪(赛默飞世尔科技有限公司);Agilent ZORBAX SB C18(2.1 mm×150 mm,1.8 μm)色谱柱,Waters Cortecs T3(2.1 mm×100 mm,1.6 μm)色谱柱;百万分之一天平(XP26,梅特勒-托利多公司)。
1.2 试剂
分析级甲醇和乙酸乙酯购自西陇科学股份有限公司;色谱级甲醇购自默克股份有限公司;色谱级甲酸和乙酸购自天津市科密欧化学试剂有限公司。
1.3 试药
牡荆苷(批号:111687-201704,含量:94.9%)、水仙苷(批号:111997-201501,含量:93.1%)、山柰酚-3-O-芸香糖苷(批号:112007-201602,含量:90.8%)、咖啡酸(批号:110885-201703,含量:99.7%)、阿魏酸(批号:110773-201614,含量:99.0%)和椴树苷(批号:112000-201802,供鉴别用)对照品均购自中国食品药品鉴定研究院;异牡荆苷(批号:ST09650120MG,含量:98.0%,上海诗丹德标准技术服务有限公司);对羟基肉桂酸(批号:JOT-10995,纯度:98.0%,四川普菲德生物技术有限公司);紫云英苷(批号:B21704,纯度:98.6%);异鼠李素-3-O-葡萄糖苷(批号:B21556,纯度:99.6%)均购自上海源叶生物科技有限公司。15批布渣叶药材经广州中医药大学中药学院黄海波教授鉴定均为椴树科植物破布叶MicrocospaniculataL.的干燥叶。取15批布渣叶药材,去除杂质,制成布渣叶饮片,按照《技术要求》制备15批布渣叶标准汤剂,其产地信息见表1所示。
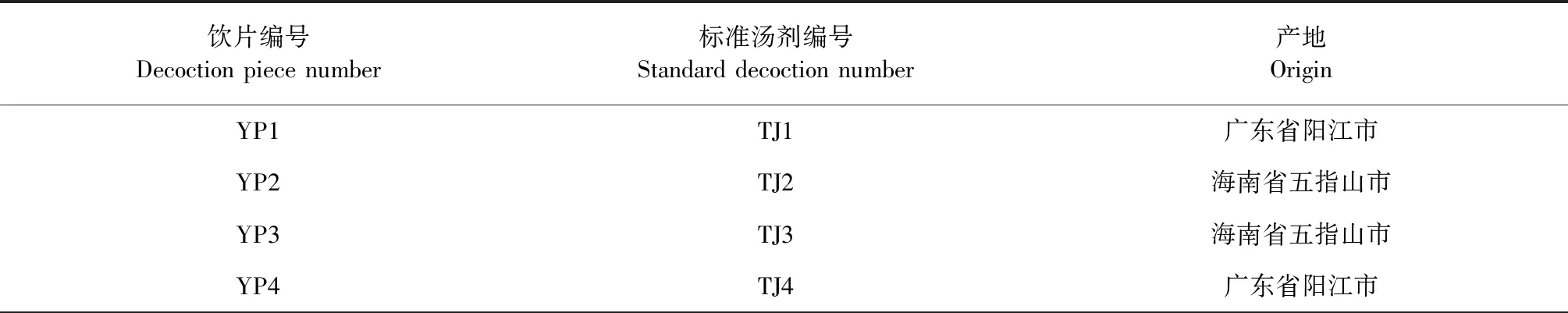
表1 15批布渣叶产地信息表
2 方法与结果
2.1 布渣叶标准汤剂的制备
取布渣叶饮片100 g,加水煎煮两次,第一次煎煮加水14倍量,浸泡30 min,武火加热煮沸后文火保持微沸30 min,趁热过滤,滤液迅速用冷水冷却;第二次煎煮加水12倍量,武火加热煮沸后文火保持微沸25 min,趁热过滤,滤液迅速用冷水冷却,合并两次煎液,减压浓缩,冷冻干燥,得布渣叶标准汤剂冻干粉。
2.2 出膏率和浸出物测定
出膏率以标准汤剂冻干粉量计,计算公式为:出膏率=(标准汤剂冻干粉总量/制备标准汤剂的饮片重量)×100%。浸出物的测定方法为:取本品2 g,精密称定,精密加入乙醇100 mL,照醇溶性浸出物(中国药典2020年版通则2201)项下的热浸法测定,结果见表2所示。15批布渣叶标准汤剂出膏率范围在9.32%~14.22%,浸出物范围在37.61%~62.52%,出膏率和浸出物数据均在均值的70%~130%或均值±3倍标准差(SD)范围内,符合《技术要求》的规定。
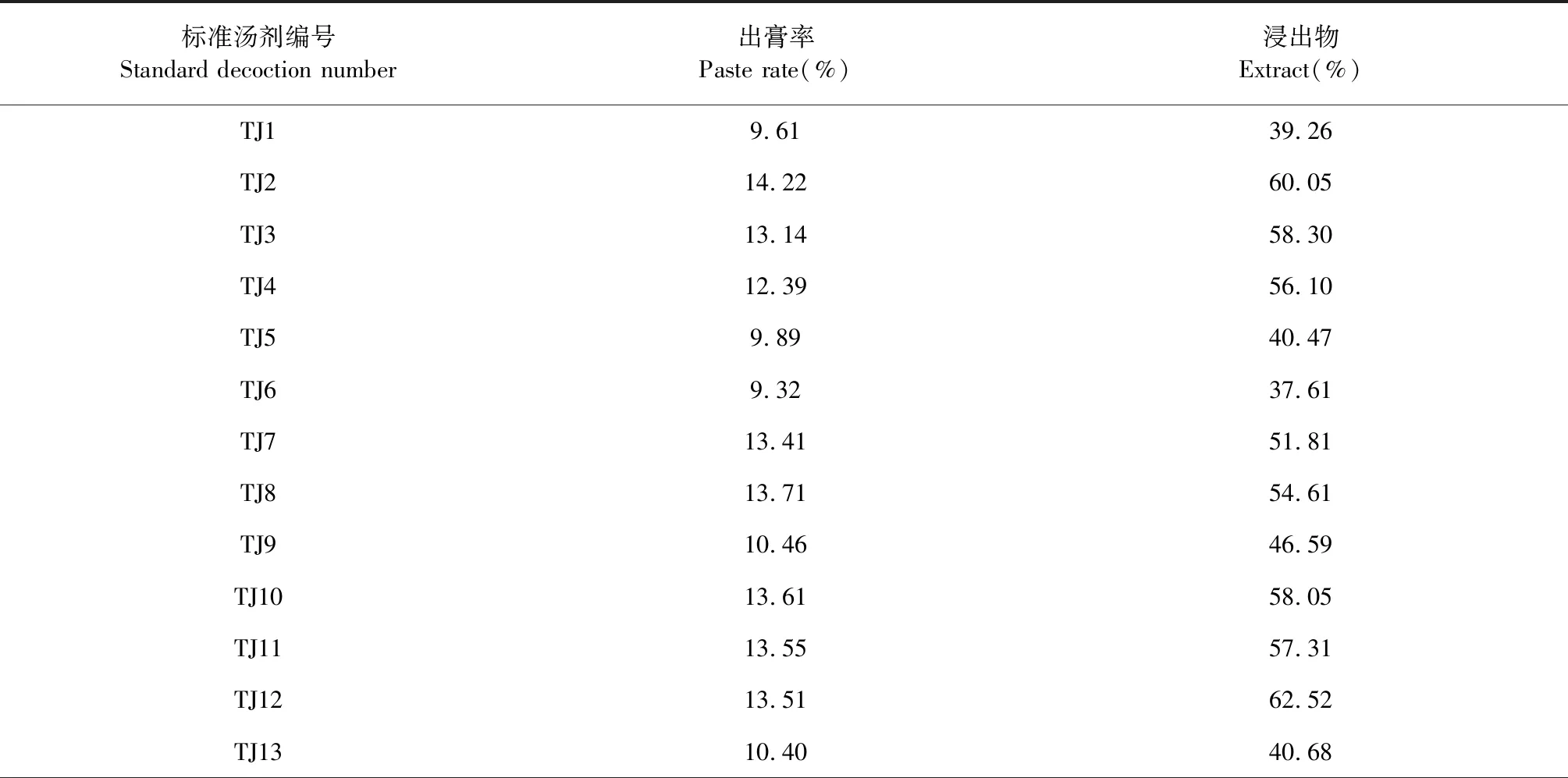
表2 15批布渣叶标准汤剂出膏率和浸出物测定结果
2.3 指纹图谱的建立
2.3.1 色谱条件
采用Agilent ZORBAX SB C18(2.1 mm×150 mm,1.8 μm)色谱柱;以甲醇为流动相A,0.1%乙酸溶液为流动相B,梯度洗脱(0~33 min,9%→17%A;33~37 min,17%→25%A;37~45 min,25%→28%A;45~60 min,28%→80%A);流速为每分钟0.30 mL;柱温为30 ℃;检测波长为315 nm;进样量为2 μL。
2.3.2 对照品溶液的制备
取咖啡酸、对羟基肉桂酸、阿魏酸、牡荆苷、异牡荆苷、山柰酚-3-O-芸香糖苷、紫云英苷、水仙苷、异鼠李素-3-O-葡萄糖苷和椴树苷对照品适量,精密称定,加甲醇制成每1 mL含咖啡酸、阿魏酸、牡荆苷、异牡荆苷各15 μg、含山柰酚-3-O-芸香糖苷、水仙苷、椴树苷各40 μg、含对羟基肉桂酸、紫云英苷和异鼠李素-3-O-葡萄糖苷各50 μg的混合溶液。
2.3.3 供试品溶液的制备
2.3.3.1 布渣叶饮片供试品溶液的制备
取本品粉末(过三号筛)约0.5 g,置具塞锥形瓶中,加50%甲醇25 mL,加热回流30 min,冷却,离心,取上清液蒸干,残渣加25mL水使溶解,用乙酸乙酯萃取3次,每次25 mL,合并乙酸乙酯液,蒸干,残渣用50%甲醇溶解,并定容到10 mL量瓶,滤过,取续滤液,即得。
2.3.3.2 布渣叶标准汤剂供试品溶液的制备
取本品适量,研细,取约0.1 g,置具塞锥形瓶中,加甲醇25 mL,超声处理(功率250 W,频率40 kHz)45 min,离心,取上清液蒸干,残渣加25 mL水使溶解,用乙酸乙酯萃取3次,每次25 mL,合并乙酸乙酯液,蒸干,残渣用甲醇溶解,定容到10 mL量瓶,过滤,取续滤液,即得。
2.3.4 指纹图谱方法学验证
2.3.4.1 精密度试验
取布渣叶标准汤剂(编号:TJ2)供试品溶液,按“2.3.1”项下色谱条件重复进样6次,以5号峰牡荆苷为参照峰S,计算其余共有峰与S峰的相对保留时间RSD值在0.04%~1.53%范围内,相对峰面积RSD值在0.34%~2.41%范围内,表明仪器精密度良好。
2.3.4.2 重复性试验
取布渣叶标准汤剂(编号:TJ2)约0.1 g,平行6份,精密称定,按“2.3.3.2”项下方法制备供试品溶液6份,采用“2.3.1”项下色谱条件进样分析,以5号峰牡荆苷为参照峰S,计算其余共有峰与S峰的相对保留时间RSD值在0.05%~0.52% 范围内,相对峰面积RSD值在 1.14%~2.84%范围内,表明该方法重复性良好。
2.3.4.3 稳定性试验
取“2.3.4.2重复性试验”项下供试品溶液,采用“2.3.1”项下色谱条件,分别在0、2、4、6、8、12、24 h进样分析,以5号峰牡荆苷为参照峰S,计算其余共有峰与S峰的相对保留时间RSD值在0.03%~0.47% 范围内,相对峰面积RSD值在0.63%~2.94%范围内,表明供试品溶液在24 h内稳定性良好。
2.3.5 指纹图谱的建立
采用“2.3”项下指纹图谱方法,对15批布渣叶饮片和标准汤剂样品进行指纹图谱测定,采用赛默飞Chromleteon 7软件将各批次样品指纹图谱以CDF格式导出,并导入《中药色谱指纹图谱相似度评价系统(2012.130723)》,分别以YP1和TJ1样品指纹图谱为参照图谱,对15批布渣叶饮片和标准汤剂指纹图谱进行多点校正和峰匹配,并生成布渣叶饮片和标准汤剂对照指纹图谱,结果见图1~3所示。结果显示,15批布渣叶饮片和标准汤剂均标识出12个保留时间相一致的共有指纹峰,表明从布渣叶饮片到标准汤剂,各指纹峰所代表的化学成分均能进行有效的转移。
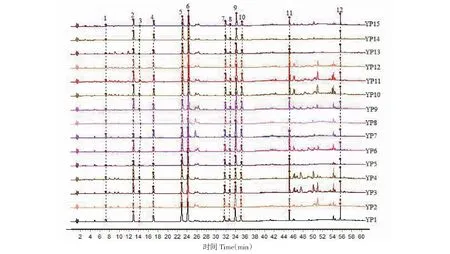
图1 15批布渣叶饮片指纹图谱Fig.1 Fingerprints of 15 batches of Microctis Folium decoction pieces
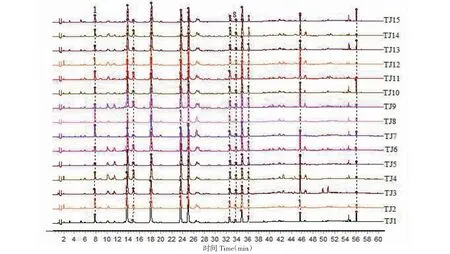
图2 15批布渣叶标准汤剂指纹图谱Fig.2 Fingerprints of 15 batches of Microctis Folium standard decoction
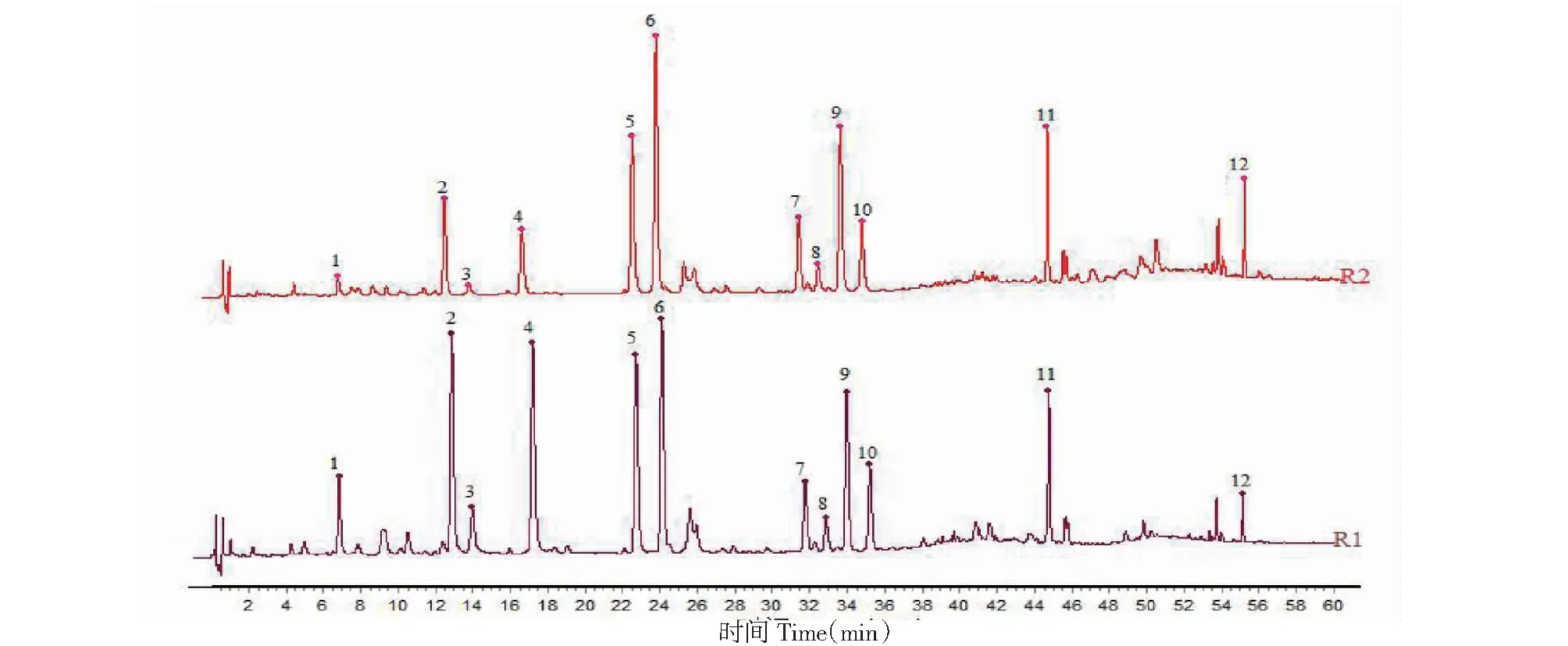
图3 布渣叶饮片和标准汤剂对照指纹图谱Fig.3 Reference fingerprints of Microctis Folium decoction pieces and standard decoction注:R1:标准汤剂对照指纹图谱,R2:饮片对照指纹图谱。Note:R1:Reference fingerprint of standard decoction;R2:Reference fingerprint of decoction pieces.
2.3.6 相似度计算
分别计算15批布渣叶饮片和标准汤剂样品指纹图谱与其对照指纹图谱的相似度,结果见表3所示,15批布渣叶饮片指纹图谱的相似度在0.957~0.995,15批布渣叶标准汤剂指纹图谱的相似度在0.960~0.995,饮片和标准汤剂指纹图谱整体相似度较高,表明指纹图谱可作为共性特征,用于布渣叶饮片和标准汤剂的质量控制。

表3 相似度计算结果
2.3.7 共有峰的指认
取布渣叶标准汤剂(编号:TJ2)供试品溶液,采用液相色谱-高分辨质谱对共有峰进行指认,色谱条件均同“2.3.1”项下;质谱采用HESI离子源,离子源参数为:鞘气流速35 Arb;辅助气流速10 arb;喷雾电压3.2 kV;S-lens电压50 V;辅助器加热温度350 ℃;毛细管加热温度350 ℃。质谱扫描参数为:正、负离子扫描模式(dd-MS2Discover);扫描范围为100~1 500m/z;一级扫描分辨率为70 000 FWHM;二级扫描分辨率为17 500 FWHM;二级碰撞能量为40 eV。分别采集供试品溶液在正、负离子模式下总离子流图(total ion chromatogram,TIC)及紫外吸收色谱图(见图4)。
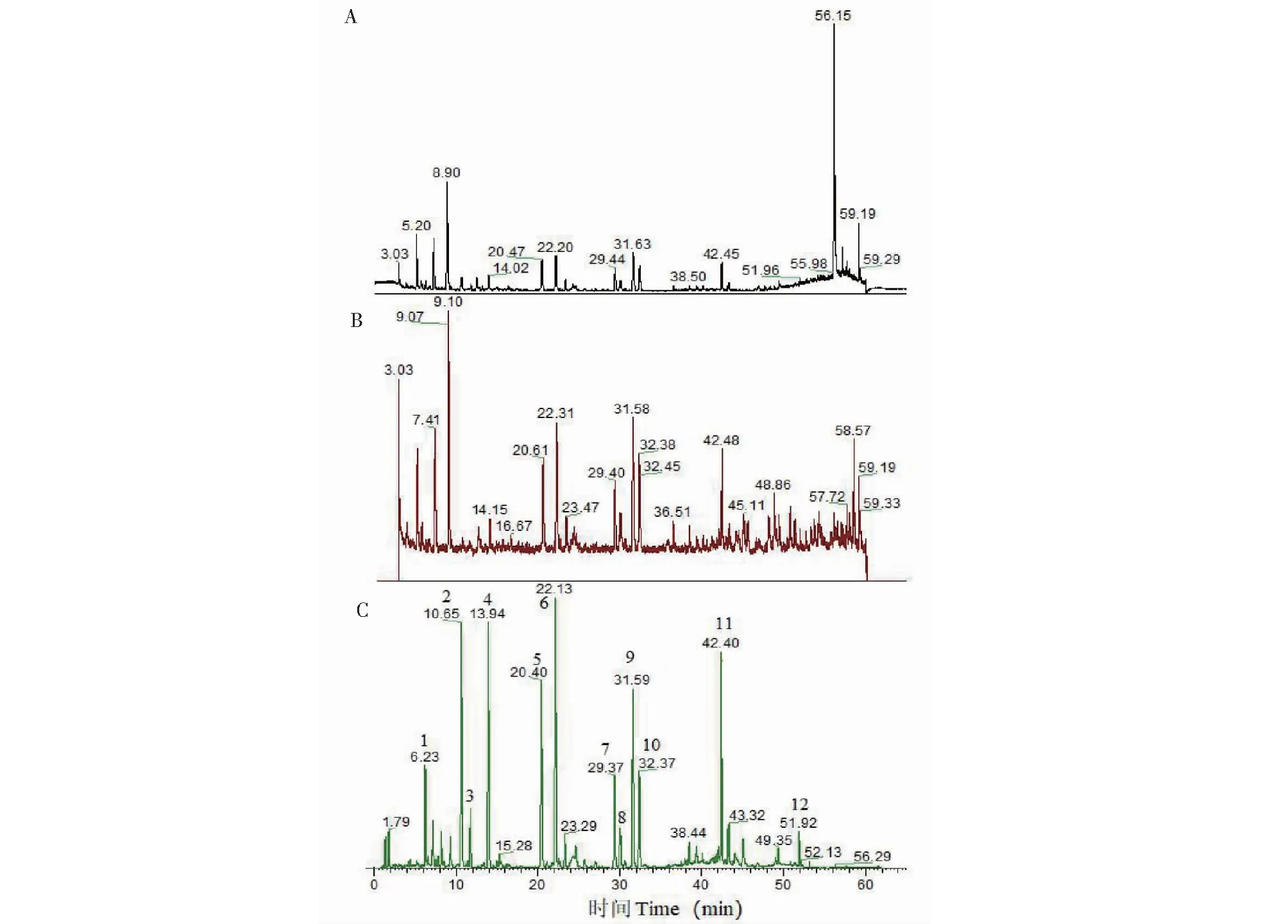
图4 布渣叶标准汤剂指纹图谱共有峰的指认Fig.4 Identification of common peaks in fingerprint of Microctis Foliumstandard decoction注:A:负离子模式下TIC;B:正离子模式下TIC;C:UV 315nm下色谱图。Note:A:TIC in negative ion mode;B:TIC in positive ion mode;C:Chromatogram at UV 315 nm.
通过化合物的精确分子量及二级质谱信息,结合本地质谱数据库资料及相关参考文献,推测峰1为咖啡酸,峰2为对羟基肉桂酸,峰4为阿魏酸,峰5为牡荆苷,峰6为异牡荆苷,峰7为山柰酚-3-O-芸香糖苷,峰8为紫云英苷,峰9为水仙苷,峰10为异鼠李素-3-O-葡萄糖苷,峰11为椴树苷。具体化合物信息见表4 所示。

表4 布渣叶标准汤剂化学成分质谱鉴定结果
2.3.8 共有峰的对照品确证
取布渣叶标准汤剂(编号:TJ3)供试品溶液和“2.3.2”项下对照品溶液,按“2.3.1”项下色谱条件进样测定,采用保留时间比对结合紫外-可见光3D光谱分析,进一步确证峰1为咖啡酸,峰2为对羟基肉桂酸,峰4为阿魏酸,峰5为牡荆苷,峰6为异牡荆苷,峰7为山柰酚-3-O-芸香糖苷,峰8为紫云英苷,峰9为水仙苷,峰10为异鼠李素-3-O-葡萄糖苷,峰11为椴树苷,结果见图5所示。
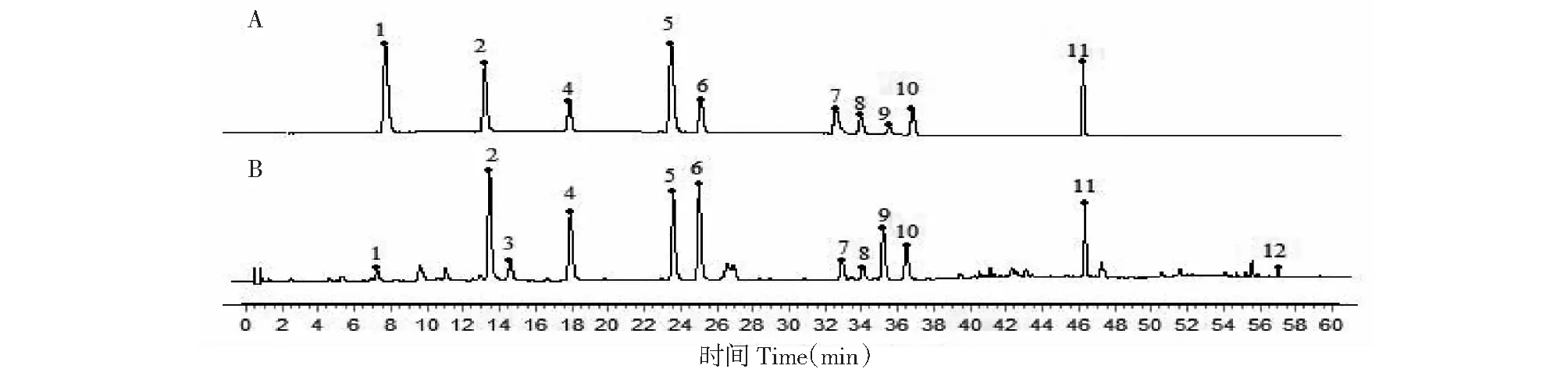
图5 共有峰的对照品确证Fig.5 Identification of reference substances for common peaks注:A:混合对照品,B:供试品。1:咖啡酸;2:对羟基肉桂酸;4:阿魏酸;5:牡荆苷;6:异牡荆苷;7:山柰酚-3-O-芸香糖苷;8:紫云英苷;9:水仙苷;10:异鼠李素-3-O-葡萄糖苷;11:椴树苷。Note:A:Mixed reference;B:Sample.1:Caffeic acid;2:4-Hydroxycinnamic acid;4:Ferulic acid;5:Vitexin;6:Isovitexin;7:Kaempferol-3-O-rutinoside;8:Astragalin;9:Narcissin;10:Isorhamnetin-3-O-glucoside;11:Tiliroside.
2.3.9 化学计量学分析
以5号峰牡荆苷为参照峰S,分别计算峰1~4、峰6~12与S峰的相对峰面积,并以相对峰面积为变量采用Simca14.1软件进行正交偏最小二乘法-判别式分析(OPLS-DA),建立OPLS-DA模型,模型经过200次置换检验,R2和Q2值右边均大于左边(见图6),证实模型有效。采用Simca14.1软件自动生成OPLS-DA得分图(见图7),15批布渣叶饮片和标准汤剂各自归为一类,且样品集中度较高。以VIP>1.0作为变量差异性筛选标准,15批布渣叶饮片和标准汤剂共筛选出5个差异标志物(见图8),以VIP大小排序,分别为峰4>峰6>峰2>峰3>峰1。分别计算15批布渣叶饮片和标准汤剂中峰1、2、3、4、6与S峰的相对峰面积,并计算15批样品的相对峰面积均值(见表5)。结果显示,从饮片到标准汤剂峰1、2、3、4的相对峰面积明显增加,而峰6的相对峰面积则明显下降。
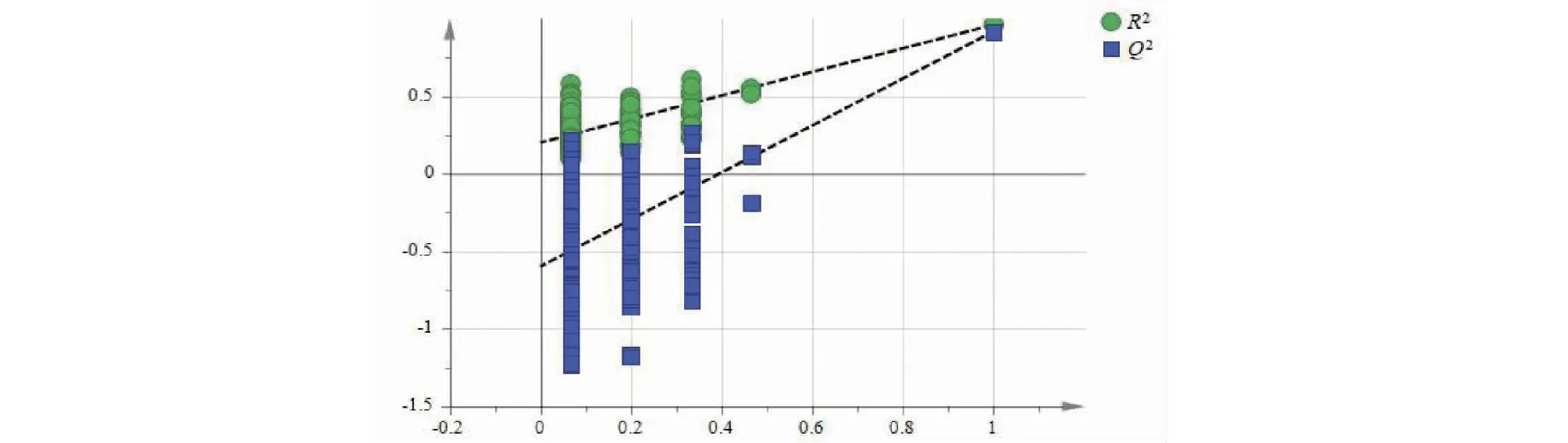
图6 OPLS-DA模型置换检验Fig.6 OPLS-DA model replacement test
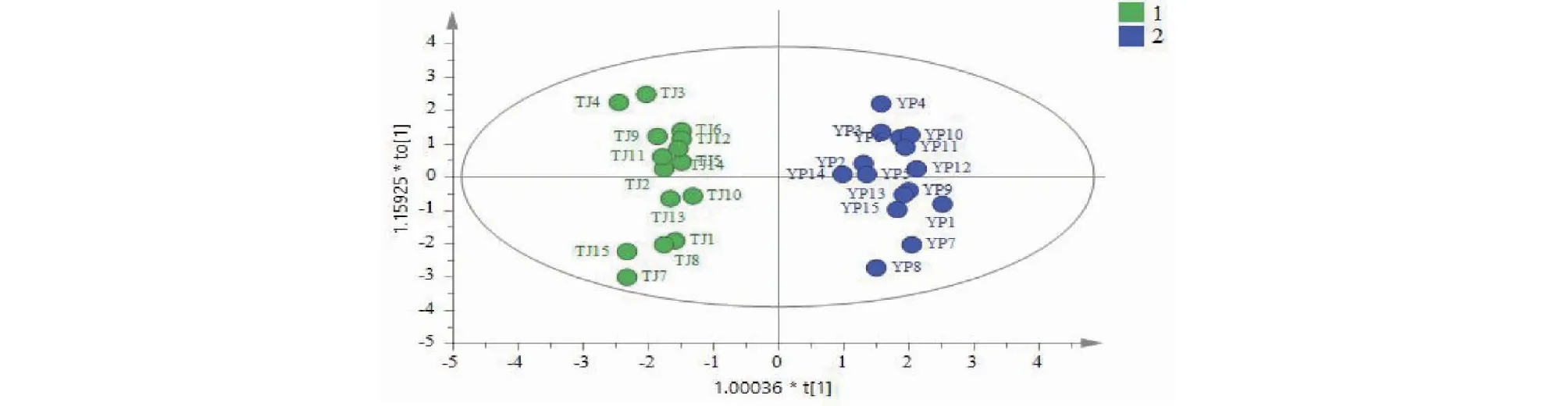
图7 OPLS-DA得分图Fig.7 OPLS-DA score plot
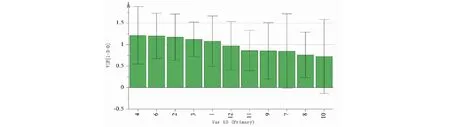
图8 VIP得分图Fig.8 VIP score plot

表5 峰1、2、3、4、6与S峰的相对峰面积均值
2.4 牡荆苷、异牡荆苷和水仙苷的含量测定
2.4.1 色谱条件
采用Waters Cortecs T3(2.1 mm×100 mm,1.6 μm)色谱柱;以甲醇为流动相A,0.1%甲酸溶液为流动相B;梯度洗脱(0~6 min,20%→25%A;6~24 min,25%→32%A;24~32 min,32%→40%A;32~35 min,40%→44%A);流速为每分钟0.35 mL;柱温为30 ℃;检测波长为339 nm,进样量为1 μL。
2.4.2 对照品溶液的制备
精密称取牡荆苷对照品2.913 mg、异牡荆苷对照品5.197 mg、水仙苷对照品16.773 mg,加70%甲醇制成每1 mL含牡荆苷13.822 μg、异牡荆苷25.465 μg、水仙苷78.078 μg的混合溶液,摇匀,即得。
2.4.3 供试品溶液的制备
2.4.3.1 布渣叶饮片供试品溶液的制备
取本品粉末(过三号筛)约2.5 g,精密称定,置具塞锥形瓶中,精密加入70%甲醇50 mL,称定重量,超声处理(250 W,频率40 kHz)1 h,取出,放冷,再称定重量,用70%甲醇补足减失的重量,摇匀,滤过,取续滤液,即得。
2.4.3.2 布渣叶标准汤剂供试品溶液的制备
取布渣叶标准汤剂适量,研细,取约0.1 g,精密称定,置具塞锥形瓶中,精密加入70%甲醇25 mL,称定重量,超声处理(功率250 W,频率40 kHz)30 min,取出,放冷,再称定重量,用70%甲醇补足减失的重量,摇匀,滤过,取续滤液,即得。
2.4.4 方法学验证
2.4.4.1 精密度考察
精密吸取“2.4.2”项下混合对照品溶液,按照“2.4.1”项下色谱条件重复进样6次,计算牡荆苷、异牡荆苷和水仙苷的色谱峰面积RSD分别为0.27%、0.57%和0.62%,表明仪器精密度良好。
2.4.4.2 重复性考察
取布渣叶标准汤剂(编号:TJ2)约0.1 g,平行6份,精密称定,按“2.4.3”项下方法制备供试品溶液6份,按“2.4.1”项下色谱条件进样测定,计算供试品溶液中牡荆苷、异牡荆苷和水仙苷的含量均值为2.761、3.290和13.920 mg/g,RSD值分别为1.83%、0.97%和0.92%,表明该方法重复性良好。
2.4.4.3 线性关系考察
精密称取牡荆苷对照品2.913 mg、异牡荆苷对照品5.197 mg、水仙苷对照品16.773 mg,置10 mL量瓶中,加70%甲醇制成每1 mL含牡荆苷276.444 μg、含异牡荆苷509.306 μg、含水仙苷1 561.566 μg的混合对照品储备液。精密量取上述混合对照品储备液0.5 mL,置2、5、10、25、50 mL量瓶中,分别加70%甲醇至刻度,摇匀,制成系列浓度对照品应用液,取上述对照品储备液和应用液,按“2.4.1”项下色谱条件依次进样分析,记录色谱峰面积。以峰面积为纵坐标(Y),对照品浓度为横坐标(X),绘制标准曲线,结果见表6所示。结果显示,3种成分的线性回归方程相关系数r均大于0.999 5,表明各成分在规定的对照品浓度范围内,峰面积与对照品浓度线性关系良好。
2.4.4.4 加样回收率试验
取牡荆苷对照品2.794 mg、异牡荆苷对照品3.627 mg、水仙苷对照品15.224 mg,置20 mL量瓶中,加甲醇制成1 mL含牡荆苷132.575 μg、异牡荆苷177.723 μg、水仙苷708.677 μg的对照品储备液。精密吸取上述对照品储液0.5、1.0、1.5 mL,平行3组,每组3份,分别置9个锥形瓶中挥干溶剂,精密加入含量已知的布渣叶标准汤剂(编号:TJ2)约0.05 g,按“2.4.3”项下方法制备供试品溶液9份,按“2.4.1”项下色谱条件测定,计算牡荆苷、异牡荆苷、水仙苷的加样回收率和RSD值,结果见表7所示。各成分的加样回收率RSD值均小于4.0%,表明该方法准确度良好。
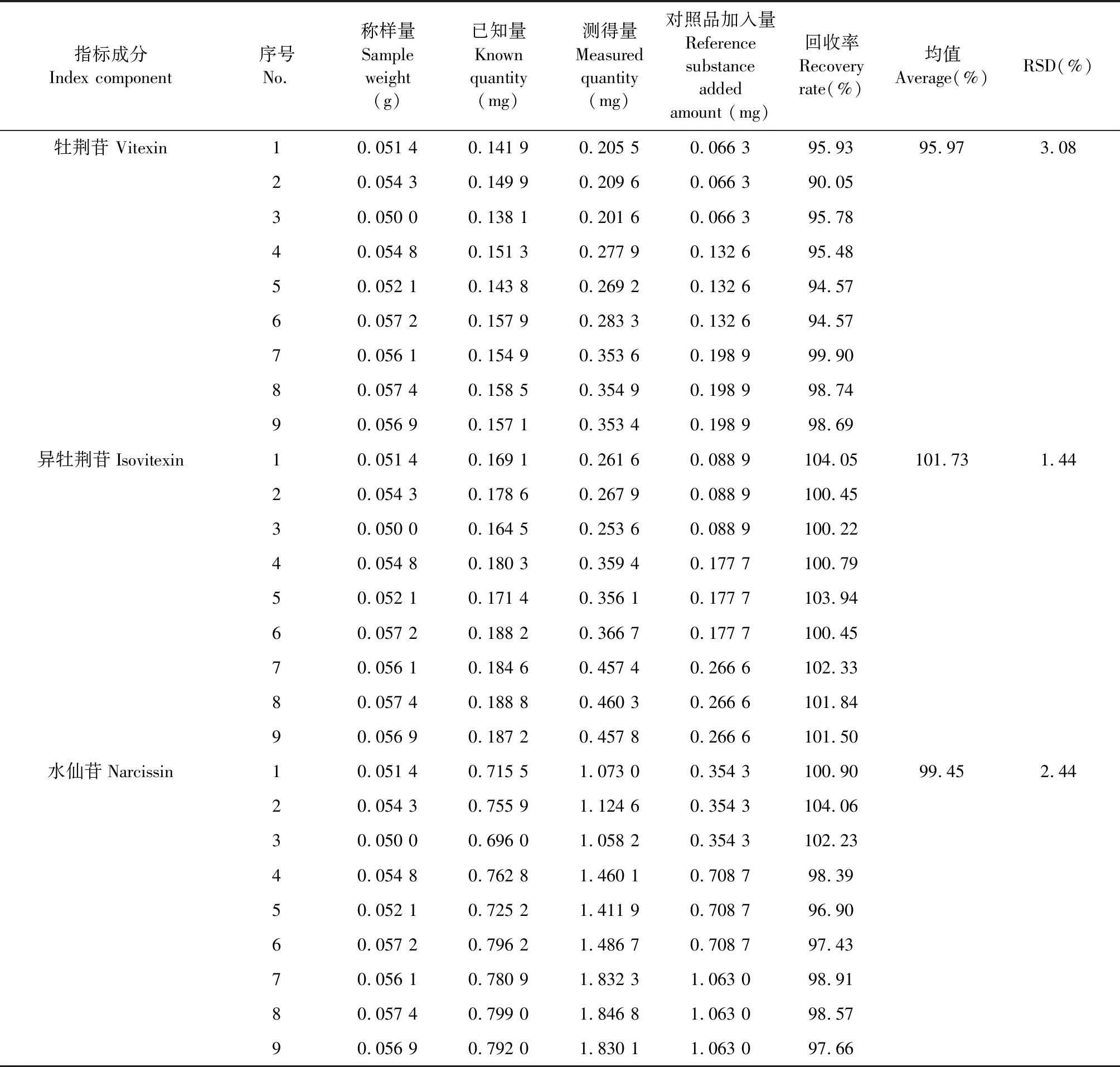
表7 各成分加样回收率测定结果
2.4.4.5 稳定性试验
精密吸取“2.3.4.2重复性试验”项下供试品溶液,按照“2.4.1”项下色谱条件,分别在0、2、4、8、12、24 h进样分析,测定供试品溶液中牡荆苷、异牡荆苷、水仙苷的峰面积,并计算峰面积RSD值分别为2.10%、0.46%和0.64%,表明供试品溶液在24 h内稳定性良好。
2.4.5 样品含量测定及转移率计算
采用“2.4.3”项下供试品溶液制备方法和“2.4.1”项下色谱条件,分别对15批布渣叶饮片和标准汤剂中牡荆苷、异牡荆苷和水仙苷的含量进行测定,并计算转移率,结果见表8所示。15批布渣叶标准汤剂牡荆苷的含量范围为1.63~4.68 mg/g,异牡荆苷的含量范围为2.16~4.25 mg/g,水仙苷的含量范围为7.86~14.18 mg/g,3种有效成分的含量均在均值±3倍标准差(SD)范围内;牡荆苷的转移率范围为41.37%~68.13%,异牡荆苷的转移率范围为31.05%~53.10%,水仙苷的转移率范围为33.11%~59.20%,3种有效成分的转移率均在均值±3倍标准差(SD)的范围内,均符合《技术要求》的规定。
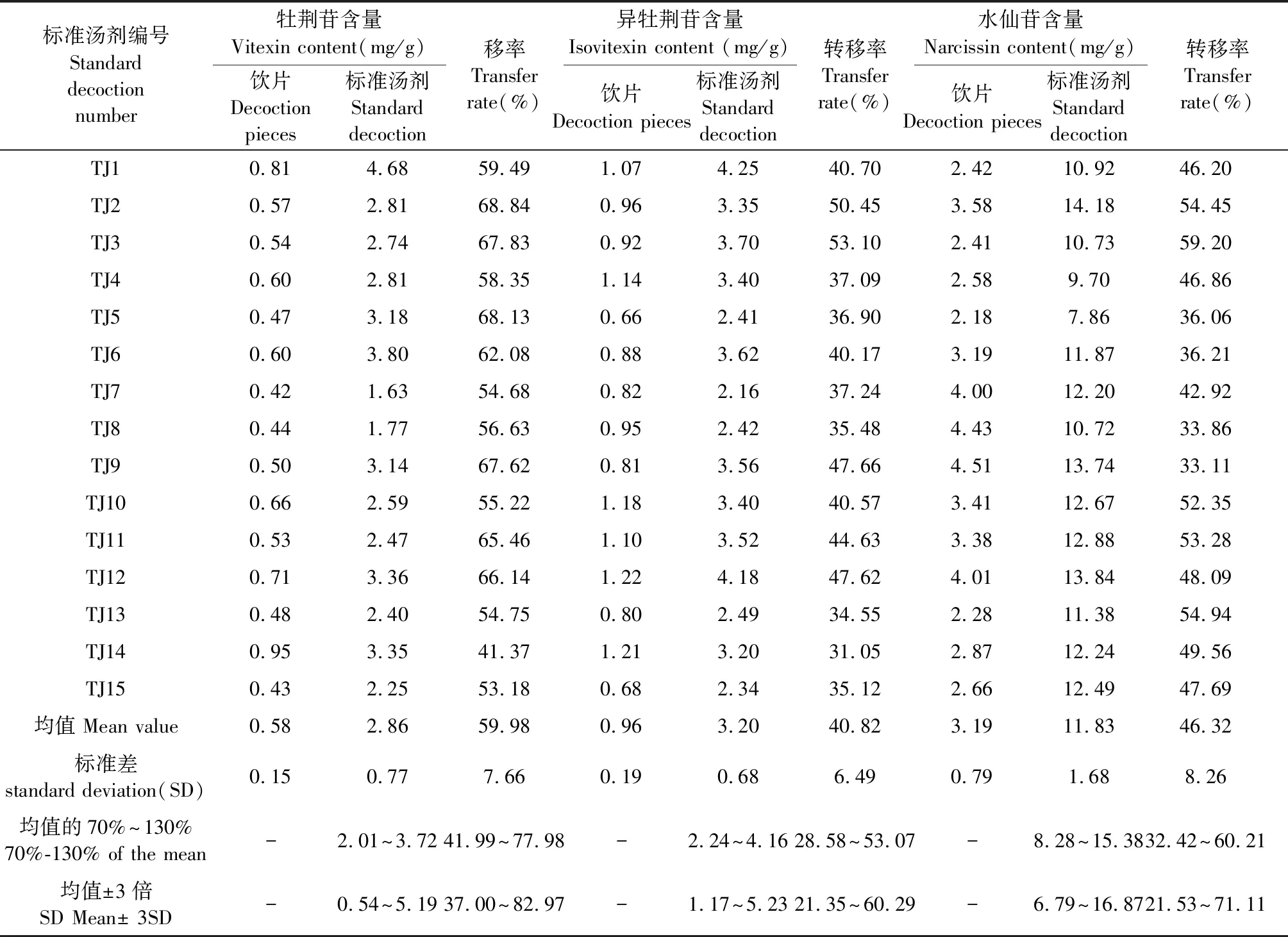
表8 15批布渣叶标准汤剂含量及转移率测定结果
3 讨论与结论
布渣叶质地较轻,但化学成分复杂,饮片煎煮过程中大量成分溢出,导致标准汤剂杂质较多,影响有效成分的色谱分离与分析,采用甲醇、乙醇等极性较大的溶剂对标准汤剂进行处理时,指纹图谱基线不平稳、色谱峰分离度较差,色谱峰杂乱,小峰较多。因此,在标准汤剂供试品前处理方法中,考察了乙酸乙酯萃取的分离纯化富集方式,减少杂质的干扰,并与常规处理方式进行对比,结果显示,采用乙酸乙酯为萃取溶剂,能够显著改善杂质峰的影响,保证主要色谱峰的纯度、分离度,解决色谱图基线不平稳,部分色谱峰吸收小的问题。在色谱条件的考察中,分别考察不同的流动相洗脱系统,如乙腈-0.1%磷酸溶液、乙腈-0.1%乙酸溶液、甲醇-0.1%甲酸溶液、甲醇-0.1%乙酸溶液等,结果显示,采用甲醇-0.1%乙酸溶液为洗脱系统,色谱峰的个数多且整体分离度较好。考虑到超高效液相色谱指纹图谱的建立,色谱柱的选择是关键,因此,本实验考察了不同粒径和品牌的色谱柱,最终选择Agilent ZORBAX SB C18色谱柱(2.1 mm×150 mm,1.8 μm),各共有指纹峰的峰形及分离度最佳。
研究显示,布渣叶含有众多黄酮类成分,Xiao等[3]采用UPLC-Q-TOF-MS/MS 技术成功从布渣叶中鉴定出31种化合物,其中28种为黄酮类,3种为酚酸类成分。药理学研究表明,黄酮类成分是布渣叶发挥消食化滞、清热利湿的重要物质基础[4-8],其代表性成分如牡荆苷、异牡荆苷和水仙苷等,在布渣叶中含量较高,且在水中具有一定的溶解性,因此,布渣叶标准汤剂以牡荆苷、异牡荆苷和水仙苷为含量测定指标,并以汤剂为基础建立布渣叶饮片的含量测定方法,从而指导标准汤剂的量值传递研究。
布渣叶药材主要分布在广东、广西和海南,属于广东产道地药材。15批布渣叶药材均采自上述3个产地,具有充分的代表性。从15批标准汤剂出膏率、浸出物以及15批布渣叶饮片3种黄酮类成分的测定结果来看,不同批次的样品存在一定的差异,最大值和最小值相差2倍左右,这种差异与药材本身质量有关,符合《技术要求》的规定,未出现明显异常值。
从转移率测定结果来看,3种黄酮类成分的转移率均值在40%~60%之间,以牡荆苷的转移率最高,其次为水仙苷和异牡荆苷,转移率整体比较稳定。指纹图谱研究结果显示,从布渣叶饮片到标准汤剂,共有峰的比例产生一定的变化, OPLS-DA分析共找到5个差异显著性色谱峰,分别为峰1、2、3、4、6,除峰3未鉴别,其余色谱峰均已知,其中峰1、2、4为酚酸类成分,因此,峰3推测也为一种酚酸类。酚酸类成分在植物中是一种特殊的存在,既能作为合成其他化合物的前体[9],也能由多种化合物水解产生[10-13],因此,布渣叶标准汤剂中酚酸类成分相对峰面积的明显增加,考虑是在标汤煎煮过程中,由其他成分水解产生。峰6异牡荆苷与S峰牡荆苷的结构类似,极性相差不大,但转移率却有较大差异,考虑标准汤剂在煎煮过程中牡荆苷与异牡荆苷存在一定的相互转化,有待于进一步研究。
本次研究以布渣叶标准汤剂的关键质量指标为基础,对布渣叶饮片标准汤剂的量值传递进行深入的研究与分析,为布渣叶配方颗粒及其相关制剂质量标准的制定提供重要参考。
猜你喜欢
基层中医药(2022年5期)2022-10-24 01:27:40
中国药学药品知识仓库(2022年5期)2022-04-11 21:25:52
世界科学技术-中医药现代化(2021年5期)2021-11-05 06:55:18
中国民间疗法(2021年14期)2021-08-30 08:24:56
中成药(2019年12期)2020-01-04 02:02:50
食品研究与开发(2019年10期)2019-05-09 09:12:34
安徽医药(2019年2期)2019-02-14 02:20:28
中国现代中药(2015年6期)2015-09-25 13:02:10
华中师范大学学报(自然科学版)(2014年2期)2014-03-28 05:11:12
中国中医药现代远程教育(2014年18期)2014-03-01 04:30:05
