
一测双评法测定咳特灵胶囊中牡荆苷与异牡荆苷的含量△
2015-09-25 13:02:10员荣孙丽丽杨立伟
中国现代中药 2015年6期
员荣,孙丽丽,杨立伟*
(1.广东药学院,广东 广州 510006;2.广东省食品药品检验所,广东 广州 510180)
·中药工业·
一测双评法测定咳特灵胶囊中牡荆苷与异牡荆苷的含量△
员荣1,2,孙丽丽2,杨立伟2*
(1.广东药学院,广东 广州 510006;2.广东省食品药品检验所,广东 广州 510180)
目的:建立咳特灵胶囊一测双评法,并进行方法学考察。方法:以牡荆苷为研究对象,建立牡荆苷与异牡荆苷的相对校正因子(RCF),并利用校正因子对异牡荆苷进行含量测定,实现一测双评;采用外标法测定牡荆苷与异牡荆苷的含量,比较计算值与实测值间的差异。结果:在一定线性范围内,牡荆苷与异牡荆苷的RCF为0.9,不同厂家的咳特灵胶囊中两个成分的计算值与实测值无明显差异。结论:本研究建立的一测双评法准确、可行,可为中药制剂多指标质量评价提供新思路。
一测双评;校正因子;高效液相色谱;牡荆苷;异牡荆苷
咳特灵胶囊现收载于《卫生部药品标准·中药成方制剂》第十四册,小叶榕干浸膏为本品的主要成分之一,其为桑科植物细叶榕FicusmicrocarpaL.f.的干燥叶加工制成的提取物。房志坚等[1]对小叶榕叶的水提物进行研究,结果表明小叶榕叶中含有牡荆苷和异牡荆苷等黄酮类化合物;李彦文[2]对小叶榕进行化学成分研究,结果表明小叶榕中含有牡荆苷和异牡荆苷。现代药理学表明:牡荆苷和异牡荆苷对金黄色葡萄球菌、表皮葡萄球菌具有较强的抑制作用,有中等的抗副流感病毒(Para 3)活性[3]。因此,测定牡荆苷和异牡荆苷的含量对于控制咳特灵胶囊质量具有重要意义。但异牡荆苷对照品价格昂贵且来源有限,因此本研究采用一测双评法[4]测定其药效成分,确定牡荆苷与异牡荆苷的内在函数关系,实现用一个对照品同步测定两个药效成分的含量[5]。一测多评理念提出之后,许多实验室及研究机构对此法在药材及制剂中的应用进行研究。目前,以一测多评法测定黄连中目标成分的含量已收载入《中华人民共和国药典》,但此法在中药制剂中的应用尚未有品种收载。
1 仪器与试药
1.1 仪器
岛津LC-20AT高效液相色谱仪系统(LC-20AT泵、CTO-15C柱温箱、SIL-20AC自动进样器、SPD-M20Ae二极阵列管检测器);KQ-300DA超声波提取器(昆山市超声仪器有限公司);纯水仪(美国Millipore公司);AG135、AEG-120G型电子天平(德国AEG工业技术公司)。
1.2 试药
牡荆苷对照品(中国食品药品检定研究院,批号:111687-220501);异牡荆苷对照品(供含量测定用,含量≥98%,上海源叶生物科技有限公司,批号:YM0506HA14)。
咳特灵胶囊由广东一力集团制药公司、广州白云山制药股份有限公司、广州白云山制药总厂、广东一力罗定制药有限公司、广州市花城制药厂提供。见表1。

表1 收集样品情况
2 方法与结果
2.1 一测双评方法学考察
2.1.1 供试品溶液的制备 取咳特灵胶囊(批号:5140303)10粒,精密称定,研细,取1.4 g,精密称定,置具塞锥形瓶中,精密加入70%甲醇50 mL,称定重量,超声处理30 min,放冷,再称定重量,用70%甲醇补足减失的重量,摇匀,滤过,取续滤液,即得。
2.1.2 对照品溶液的制备 精密量取牡荆苷、异牡荆苷对照品适量,用70%甲醇溶液配成质量浓度分别为10.61、14.40 μg·mL-1的牡荆苷和异牡荆苷对照品溶液。
2.1.3 阴性对照品溶液的制备 取小叶榕干浸膏阴性样品1.4 g,照供试品溶液的制备方法,制成阴性对照溶液。
2.1.4 色谱条件 以十八烷基硅烷键合硅胶为填充剂;以甲醇为流动相A,0.05%磷酸溶液为流动相B,按表2进行梯度洗脱;检测波长为335 nm;流速为1.0 mL·min-1;柱温为25 ℃;进样量为10 μL。理论板数按牡荆苷峰计算应不得低于3000。在上述色谱条件下,各组分分离度较好,见图1。

表2 拟定的流动相梯度表
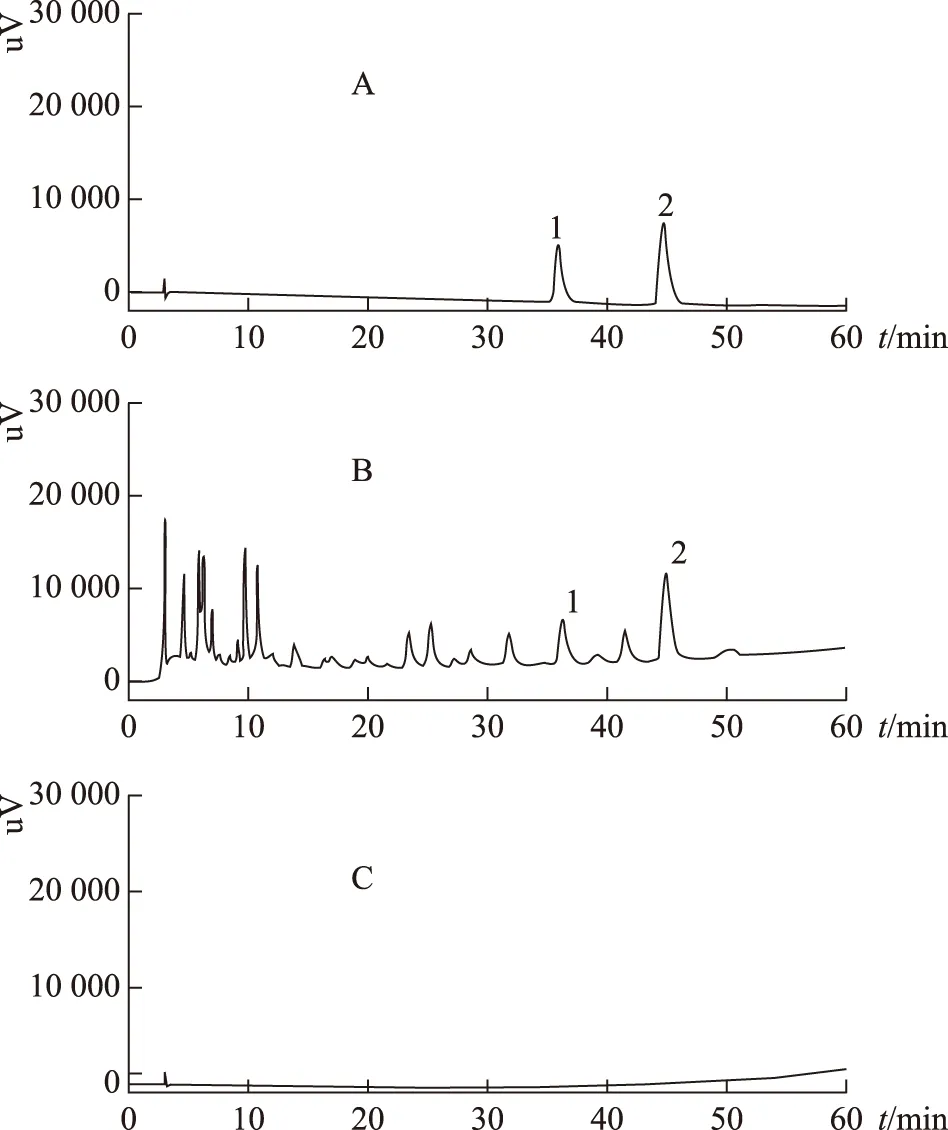
A.对照品;B.样品;C.阴性对照品;1.牡荆苷;2.异牡荆苷。图1 咳特灵胶囊样品及对照品色谱图
2.1.5 线性考察 精密称取牡荆苷对照品8.31 mg,置100 mL量瓶中,加70%甲醇溶解并稀释至刻度,得质量浓度为0.083 1 mg·mL-1的牡荆苷储备液;精密称取异牡荆苷对照品8.40 mg,置100 mL量瓶中,加70%甲醇溶解并稀释至刻度,得质量浓度为0.084 0 mg·mL-1的异牡荆苷储备液。取上述牡荆苷、异牡荆苷对照品储备液各2 mL,加入25 mL容量瓶中,加70%甲醇溶解并稀释至刻度,得质量浓度分别为0.006 648、0.006 720 mg·mL-1的牡荆苷、异牡荆苷混合对照品溶液。分别进样5、10、40、60、100 μL。分别以牡荆苷和异牡荆苷浓度为横坐标,峰面积为纵坐标绘制标准曲线,得线性方程,异牡荆苷:Y=2.783×107X-1800,r=0.999 6;牡荆苷:Y=2.573×107X-1.176×105,r=0.999 8,结果表明牡荆苷和异牡荆苷分别在0.033 2~0.636 6 μg和0.033 6~0.672 0 μg线性关系良好。
2.1.6 校正因子的计算 以牡荆苷为内标,计算牡荆苷对异牡荆苷的相对校正因子,计算公式:

式中A为组分峰面积,C为组分浓度,i为内标物,S为其他组分,计算结果见表3。
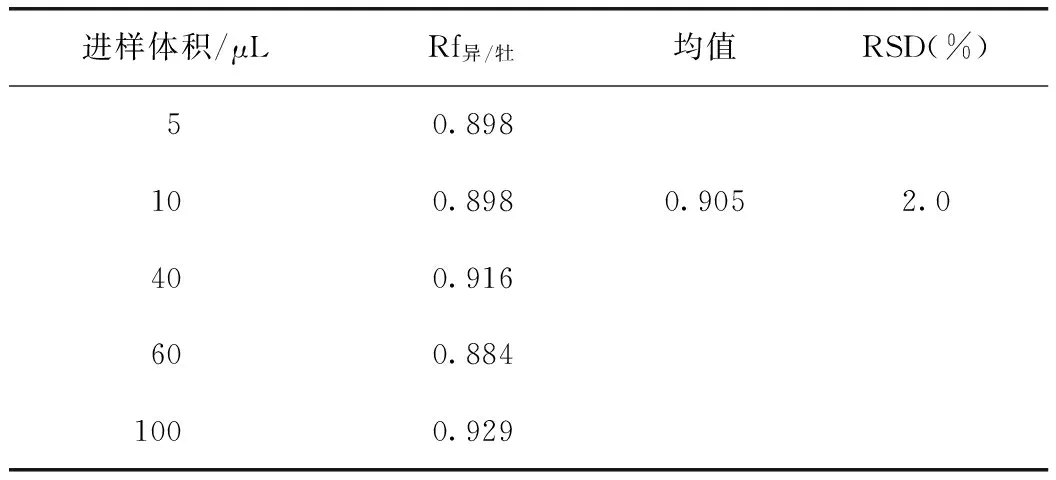
表3 相对校正因子的计算
2.1.7 重复性试验 取同一供试品(批号:5140303),精密称取6份,按拟定方法测定,结果表明牡荆苷的平均质量分数为0.260 3 mg·g-1,RSD为1.5%;异牡荆苷的平均质量分数为0.580 8 mg·g-1,RSD为2.3%,说明本方法的重复性良好。
2.1.8 稳定性 精密吸取同一供试品(批号:5140303)溶液,分别在1、3、6、9、12、16、24 h,注入高效液相色谱仪,记录峰面积。结果牡荆苷峰面积的RSD为2.0%,异牡荆苷峰面积的RSD为2.5%,表明供试品溶液在24 h内基本稳定。
2.1.9 回收率考察 分别取已测得含量的供试品(批号:5140303,牡荆苷0.260 3 mg·g-1,异牡荆苷:0.580 8 mg·g-1)0.4、0.7、1.4 g各4份,精密称定,分别加入2.1.2项下的牡荆苷对照品储备液1、1.5、3 mL和异牡荆苷对照品溶液2、3、6 mL,按2.1.1项下方法制备溶液,按拟定的方法进行测定,记录色谱峰峰面积并计算回收率,结果表明,本方法对牡荆苷及异牡荆苷含量测定准确性较好。详见表4~5。
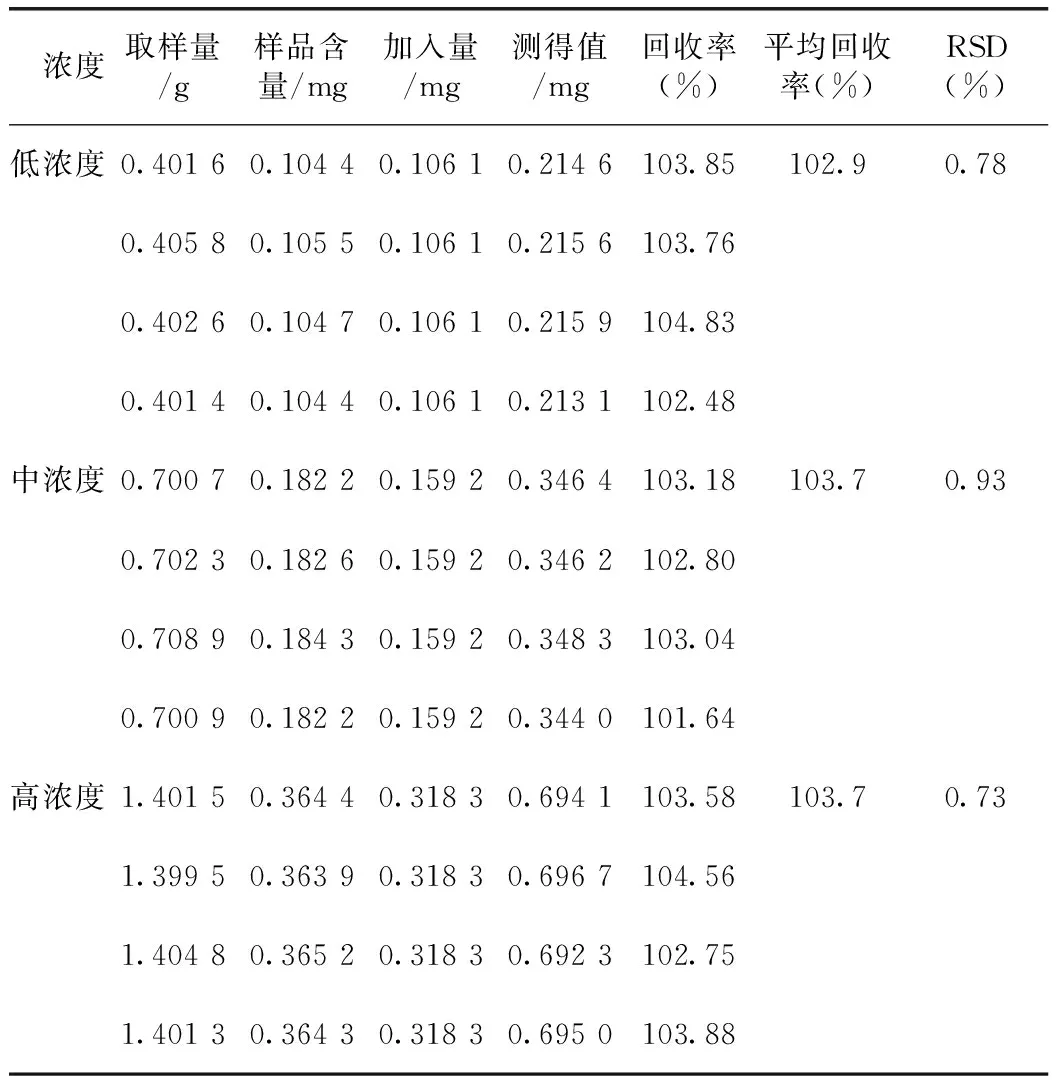
表4 牡荆苷回收率结果
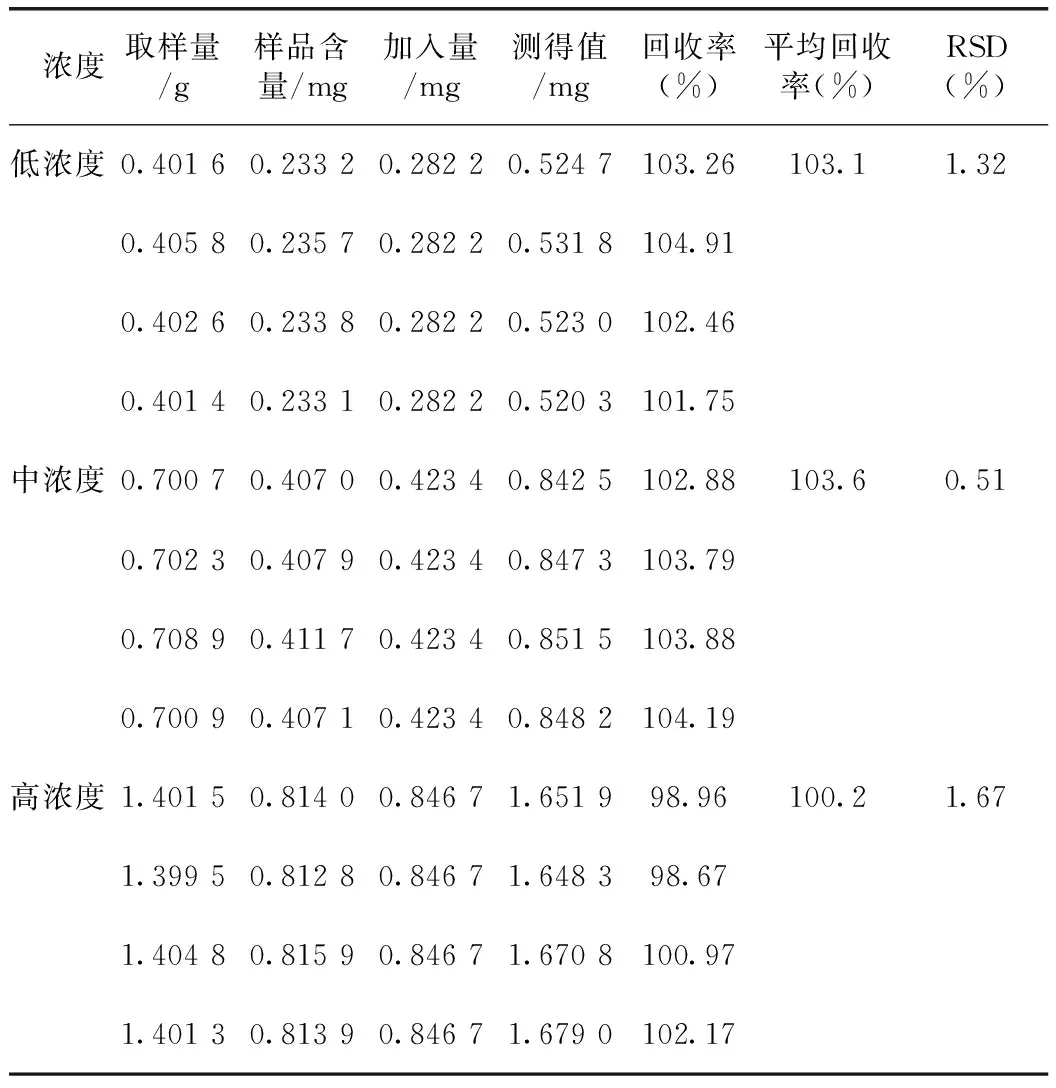
表5 异牡荆苷回收率结果
2.2 校正因子重现性考察
2.2.1 色谱柱及高效液相色谱仪考察 取2.1.5项下混合对照品溶液,进样5、10、40、60、100 μL,按2.1.6项下方法计算异牡荆苷的相对校正因子。
本试验考察了Waters 2695、岛津LC-2010A HT两种高效液相色谱仪和Welch XB-C18(250 mm×4.6 mm,5 μm)、Purospher STAR RP-18 endcapped(250 mm×4.6 mm,5μm)、Sepox Bio-C-18(250 mm×4.6 mm,5 μm)3种色谱柱,结果见表6。
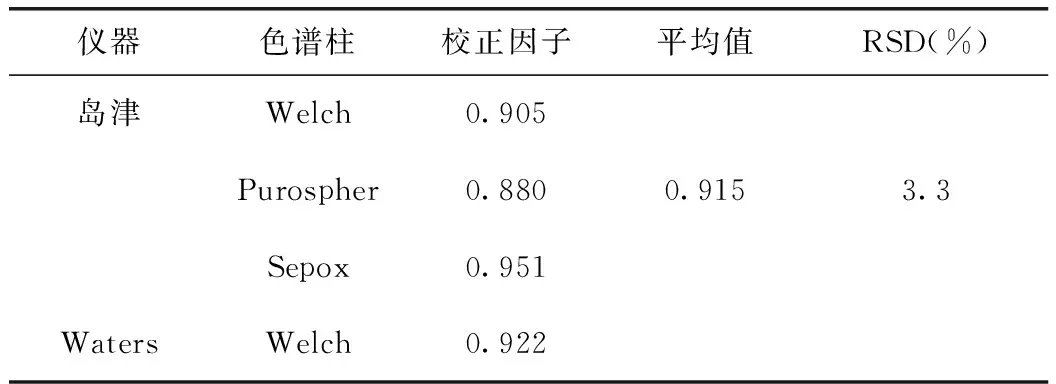
表6 不同仪器及色谱柱的相对校正因子比较
2.2.2 实验室考察 采用一测双评法,经两个实验室进行复核试验。实验室1所得结果为0.893,实验室2所得结果为0.905。
2.2.3 待测组分色谱峰定位 利用相对保留时间定位,分别考察了不同柱温、不同色谱柱及不同仪器等条件下,异牡荆苷峰相对于牡荆苷峰的保留时间。结果表明,在以上不同条件下,保留时间比较稳定,说明以相对保留时间定位是可行的,结果见表7。
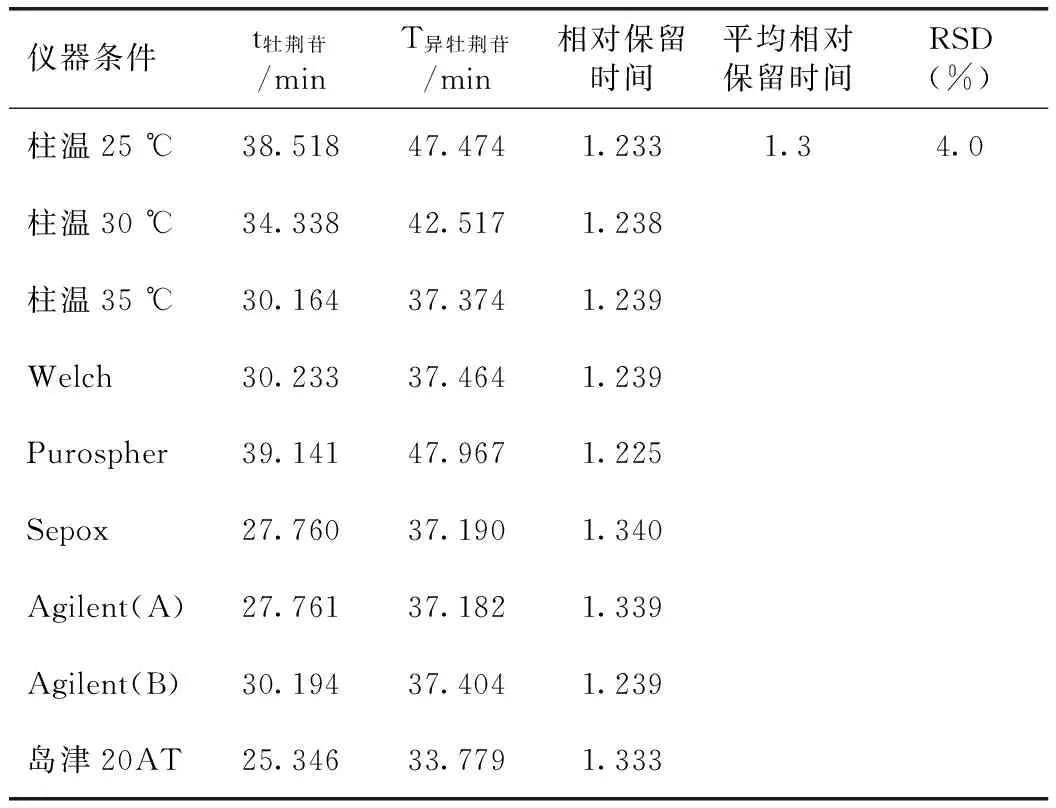
表7 相对保留时间的考察
2.3 一测双评法与外标法结果比较
对收集的24批样品分别按照外标法和一测双评法进行测定,结果异牡荆苷的含量结果相对偏差均不超过2.2%。说明本研究选择Rf异/牡为0.90是合理的,对测定结果的影响不大,见表8。
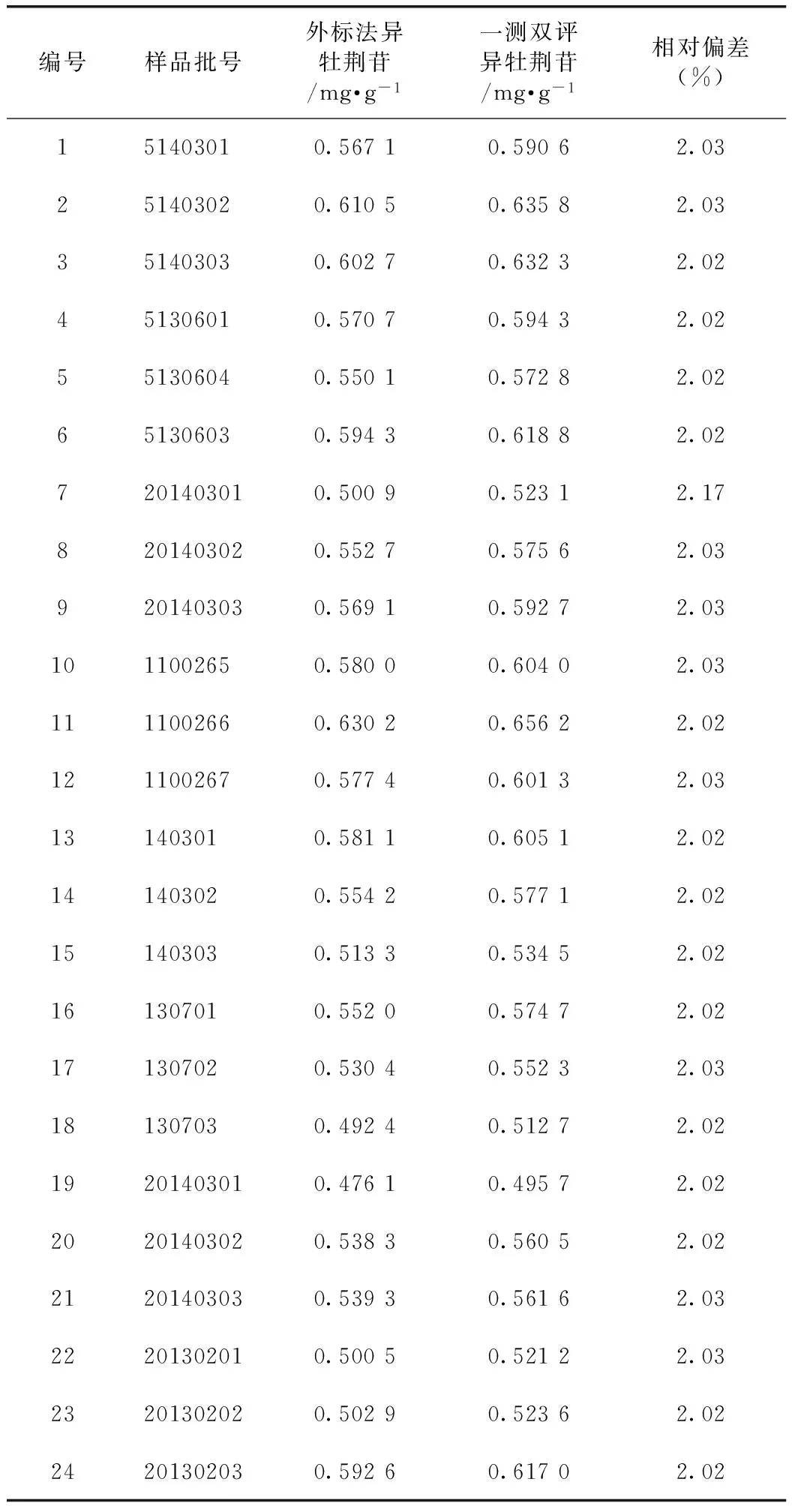
表8 两种方法测定异牡荆苷的比较
2.4 含量测定
按拟定的含量测定方法对收集的5个生产厂家24批样品分别进行测定,结果见表9。24批样品中牡荆苷和异牡荆苷含量之和的均值为0.837 mg·g-1。
2.5 限度制订
所测的24批样品中牡荆苷和异牡荆苷含量之和的均值为0.837 mg·g-1,即0.42 mg/粒(每粒胶囊0.5 g,分别称取20粒,记录其各自总重量,然后取出内容物,再称量胶囊壳的重量,然后计算每粒的净重量,并求得平均值)。由于小叶榕药材的质量标准中无牡荆苷和异牡荆苷含量测定项,故拟定本品含量测定限度以所收集到的样品测定结果的平均值(0.42 mg/粒)为依据。考虑到药材来源及大生产等可能产生的差异,笔者将所测定结果的平均值下调30%,拟定本品限度。咳特灵胶囊含小叶榕浸膏,以牡荆苷(C21H20O10)和异牡荆苷(C21H20O10)的总量计,不得少于0.28 mg/粒。
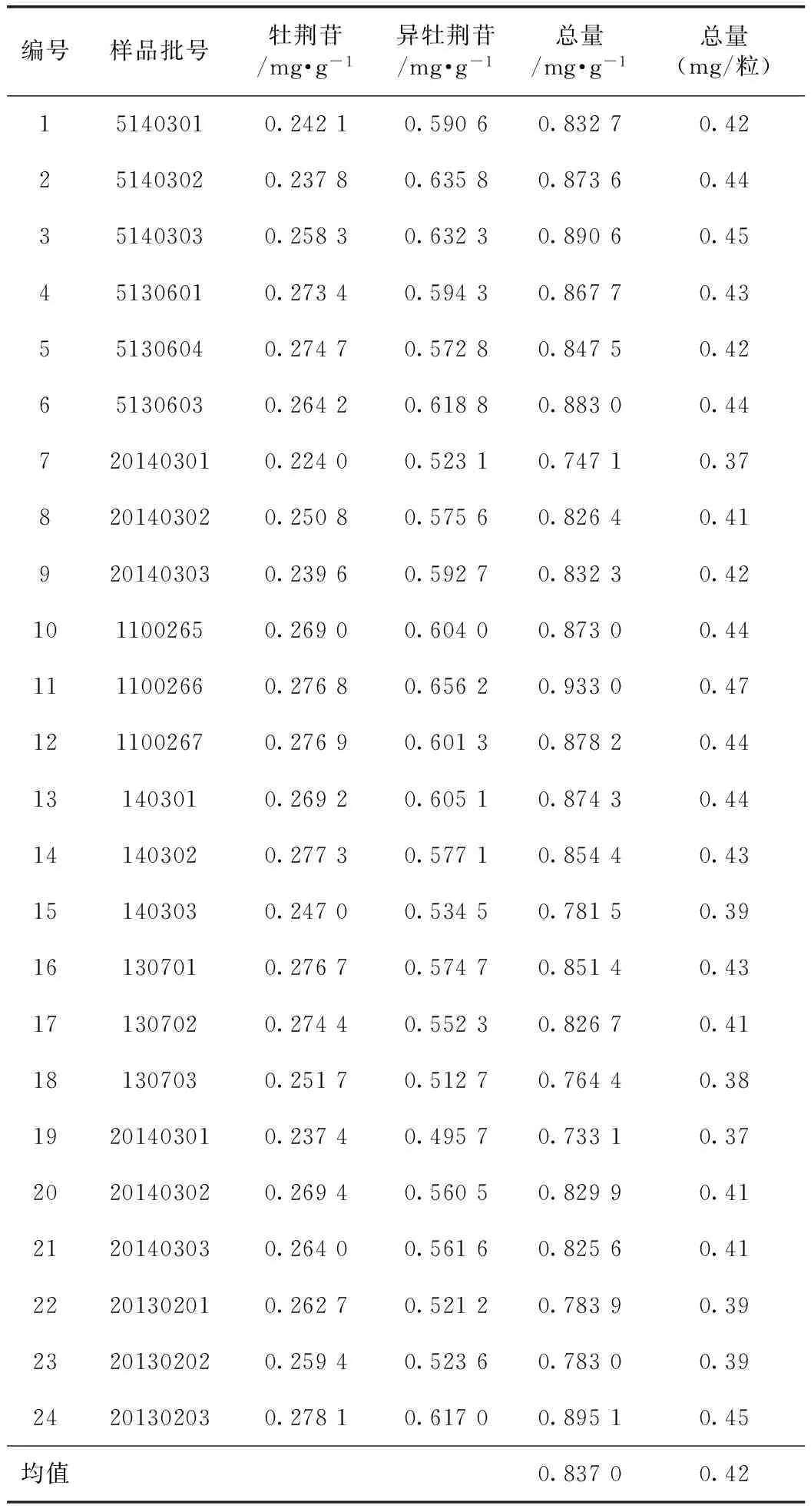
表9 供试品含量测定结果
3 讨论
3.1本试验比较了不同提取溶剂、提取方式、提取时间、溶剂用量等,最后确定以70%甲醇25 mL超声30 min,此条件下提取较为完全,且方法简便。
3.2牡荆苷与异牡荆苷均为咳特灵胶囊的药效成分,异牡荆苷价格昂贵,较难得到,且牡荆苷与异牡荆苷为同分异构体,因此以牡荆苷为内参物计算异牡荆苷的含量是合理的。
3.3 创新点
在进行一测双评研究时,笔者建议尽量使各物质的浓度一致,以减少各对照品浓度不一致造成的误差。本研究,配成浓度比为1∶1的混合对照品,在保证两者浓度相同的情况下,相对校正因子仍与理论值1差别较大,因此,相对校正因子与物质结构的关系有待进一步研究;同时建议在进行一测多评计算相对校正因子时,将各对照品的浓度比配成1,此项建议并未见此领域研究者提出。
3.4对于药物的整体质量评价,多采用指纹图谱法,而一测双评法是在药效成分明确基础上的定量分析。目前,一测双评法多用于药材中药效成分的研究,已有很多文献报道在其中药制剂中的应用,《中华人民共和国药典》中尚无关于中药成药的记载。
[1] 房志坚,戴臻,李书渊.小叶榕叶HPLC指纹图谱的研究[J].中药材,2008(10):31-35.
[2] 李彦文.小叶榕化学成分和质量标准研究[D].北京:北京中医药大学,2008.
[3] 张雪,徐道华.牡荆苷的药理作用研究进展[J].中国医药导报,2013(35):35-38.
[4] 王智民,钱忠直,张启伟,等.一测多评法建立的技术指南[J].中国中药杂志,2011(6):657-658.
[5] 陈建伟,李祥,步达,等.一测多评法及其在中药材质量控制中的应用[C].//中华中医学会中药分析分会第五届学术交流会论文集.沈阳:中华中医药学会中药分析分会,2012:20-23.
SimultaneousDeterminationofVitexinandIsovitexinofKetelingCapsulesbyDouble-ComponentsAssaywithSingleMarker
YUANRong1,2,SUNLili2,YANGLiwei2*
(1.GuangdongPharmaceuticalUniversity,Guangzhou510006,China;2.GuangdongInstituteforFoodandDrugControl,Guangzhou510180,China)
Objective:To establish a quantitative assay of double-components by single-marker for determination of two components in Keteling capsules and and conduct the method validation test.Methods:Vitexin was selected as an internal reference substance,RCF of isovitexin was calculated.The contents of two components were determined by both external standard method and Double-Components Assay by Single Marker.The validity of the QAMS method was evaluated by comparison of their quantitative results of both methods.Results:RCF of isovitexin with reference to vitexin were 0.90 and the quantitative results of both external standard method and QAMS had no significant difference.Conclusion:The established method is accurate and feasible,and can provide a new mind for the quality assess of preparation of Chinese traditional medicine.
QAMS;RCF;HPLC;Vitexin;Isovitexin
10.13313/j.issn.1673-4890.2015.6.018
2014-11-26)
国家自然科学基金资助项目(81102146)
*
杨立伟,主任药师,博士,研究方向:中药质量控制与快速检测;E-mail:yangliwei33@aliyun.com
猜你喜欢
High Technology Letters(2021年4期)2022-01-09 02:08:16
机电信息(2021年10期)2021-09-10 07:22:44
河北果树(2020年4期)2020-11-26 06:05:00
机电信息(2020年34期)2020-01-21 05:58:46
食品研究与开发(2019年10期)2019-05-09 09:12:34
海峡姐妹(2019年1期)2019-03-23 02:42:40
安徽医药(2019年2期)2019-02-14 02:20:28
中成药(2018年9期)2018-10-09 07:18:46
机电信息(2015年28期)2015-02-27 15:58:02
华中师范大学学报(自然科学版)(2014年2期)2014-03-28 05:11:12
