
OBSCN突变对于肝癌细胞增殖和迁移能力的影响
2023-09-13 01:47付琳琳赵雪梅
山东第一医科大学(山东省医学科学院)学报 2023年7期
付琳琳 王 玉 赵雪梅
1. 山东第一医科大学(山东省医学科学院)药学院,山东 泰安 271016;2. 山东第一医科大学(山东省医学科学院)临床与基础医学院,山东 济南 250117
细胞骨架蛋白Obscurin(由OBSCN编码的蛋白)[1]是细胞内的分子支架,广泛分布于横纹肌、脑、肝、脾和肺等多种组织细胞[2]。有研究表明,OBSCN基因在乳腺癌和结直肠癌中表现了高度突变[3-4]。在乳腺癌患者样本中也观察到OBSCN基因突变率超过了15%[5]。原发性肝癌是我国目前发病率高、危害性大的恶性肿瘤之一[6-7],临床有效治疗手段缺失[8-9]。本研究以人肝癌细胞HepG2为模型,构建过表达对照空载体HepG2 GL119细胞系、OBSCN过表达HepG2 H21157 细胞系、敲除对照空载体HepG2 ecas 细胞系和OBSCN敲除HepG2 OBSCN KO 细胞系,初步探究OBSCN突变对于肝癌细胞增殖和迁移能力的影响,为进一步探究OBSCN基因在肝癌发生发展中所起到的作用奠定了基础,为改善肝癌预后不良的现状提供了新的治疗思路。
1 材料与方法
1.1 材料
人肝癌细胞HepG2、MEM 培养基(武汉普诺赛生命科技有限公司),NonEssentialAmino、丙酮酸钠(美国GIBCO 公司),DMEM 培养基、胰蛋白酶消化液(山东思科捷生物技术有限公司),Polybrene(美国Sigma 公司),过表达对照病毒GL119(pSLenti-CMV-MCS-3xFLAG-PGK-Puro-WPRE)、目 的 病 毒H21157[pSLenti-CMV-OBSCN(3 250 ~ 4 749bp,G1333R)-3xFLAG-PGK-Puro-WPRE](上海和元生物技术有限公司),DNA 引物合成与克隆质粒的序列测定(上海生工生物工程技术服务有限公司),BsmBI-v2(美国NEB 公司),Lipofectamine™ 2000 转染试剂(美国invitrogen 公司),嘌呤霉素(Solarbio),rTag PCR Mix、DNA琼脂糖凝胶纯化试剂盒、无内毒素质粒小提试剂盒(北京天根生化科技有限公司),OBSCN 抗体(Sigma),CCK-8 试剂盒(上海碧云天生物技术有限公司),transwell小室(美国康宁公司)。
1.2 方法
1.2.1 荧光定量PCR 实验所用细胞均为处于对数生长期细胞,于37℃,5% CO2培养箱中培养,完全培养基配比为DMEM/MEM 培养基∶FBS 胎牛血清∶青链霉素= 9∶1∶0.1,细胞铺板前使用台盼蓝染液进行细胞计数。提取细胞RNA,反转录得到cDNA,设计合成引物(表1)。荧光定量PCR(qPCR)混悬体系上机检测,验证OBSCN有无本底表达。
1.2.2 慢病毒滴度测定与慢病毒感染 取处于对数生长期的HepG2细胞,在24孔板中以2.5 × 105/孔的浓度接种,将慢病毒H21157进行梯度稀释,共做3 个梯度,即每孔含10、1、0.1 μL 病毒,加至接种好细胞的24 孔板中,感染12~20 h 后,更换新鲜培养基。感染72 h 后收集细胞并进行慢病毒滴度的测定。
慢病毒滴度测定公式为慢病毒滴度(IU/mL) =(C × N × D × 1 000)/V,其中:V = 加入的稀释病毒的体积数,C = 平均每基因组整合的病毒拷贝数,N = 感染时细胞的数目,D = 病毒载体的稀释倍数。
将HepG2 细胞以4 × 105/孔的浓度接种于6 孔板,12~20 h 后感染病毒,每孔加10 μg polybrene。感染12~20 h后弃去培养基,每孔加入2 mL新鲜的培养基。72 h后加入终浓度为2 μg/mL puromycin 的新鲜培养基。每隔2~3 天换1 次终浓度为2 μg/mL puromycin 的新鲜培养基,筛选2 周后,扩大培养,得到OBSCN 过表达细胞系,其中空载体对照HepG2 GL119稳转细胞系感染的病毒载体为HepG2-pSLenti-CMV-MCS-3xFLAG-PGK-Puro-WPRE,OBSCN过 表达HepG2 H21157 稳转细胞系感染的病毒载体为:HepG2-pSLenti-CMV-OBSCN(3 250 ~ 4 749 bp,G1333R)-3xFLAG-PGK-Puro-WPRE。
1.2.3 引导RNA的设计 以人类基因组全序列数据库的数据(https://www. ncbi. nlm. nih. gov/)和Crispr Cas9 慢病毒质粒LentiCRISPR v2 中U6 启动子的转录偏好要求,选取染色体序列中原始OBSCN的外显子位点(3 951 ~ 3 972 位),设计包含BsmBI核酸内切酶酶切产生的粘性末端对应序列的引导RNA(guiding RNA, gRNA)正反引物OBSCN-exon gsRNAF 和OBSCN-exon gsRNAR,见表2。同时参照载体LentiCRISPR v2 的全序列,设计针对两BsmBI 酶切位点上下游2 160 bp 的鉴定引物BsmBI ID F和BsmBI ID R,见表2。
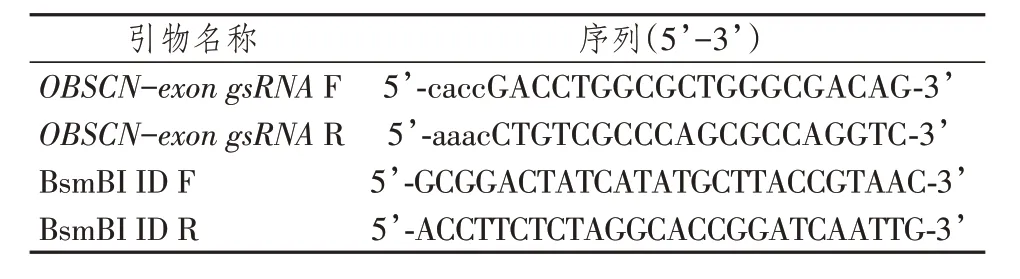
表2 引导RNA和鉴定引物序列
1.2.4 构建重组质粒reOBSCN 将含引物OBSCN-exon gsRNAF/R 的聚合酶链式反应(PCR)反应管按照37℃ 30 min,95℃ 5 min,5℃/min降温至25℃的反应条件运行退火反应。PCR退火反应结束后,使用0.8%的琼脂糖凝胶板进行电泳分离。
经UV瞬时照射、切取目的条带进行纯化回收,回收产物定量后,按照100 ng经BsmBI酶切线性化的载体质粒LentiCRISPR v2、1 μL 1∶10 稀释后的上一步反应液、5 μL 10XNEBuffer、2 μL BsmBI、5 μL ATP、5 μg BSA、2.14 μL T4 DNA Ligase和ddH2O,将反应液补足至50 μL的反应体系配制连接反应液;在充分混匀后,按照37℃ 5 min,20℃ 5 min,循环1~20 次;80℃ 20 min循环21次反应条件进行连接反应。待反应管充分冷却后,将其转移至-20℃冷冻保存。
取5 μL连接反应液,按分子克隆[10]建议的方案转化DH5α感受态细菌,涂布含氨苄青霉素的LB固体平板培养后,转移至37℃温箱培养16~24 h 后,取出平板培养基观察、标记优势抗性生长的单菌落。用无菌的10 μL枪头依次轻缓地挑取各优势抗性生长的单菌落,分别转移至含引物BsmBI ID F 和BsmBI ID R 的PCR 反应液中,其中细菌的体积按0 μL计,进行PCR扩增[11]和电泳检测。
1.2.5 质粒提取与鉴定 用无菌枪头挑取PCR初步筛选阳性的单菌落,转移至分装有6 mL含氨苄青霉素LB 培养基的试管中,在37℃温浴摇床震荡培养16 h。取1 mL菌液送检测序;同时将剩余菌液用无内素质粒提取试剂盒提取质粒。质粒溶液经Nanophotometer 测定浓度及纯度后,取1 μg 转移至含引物BsmBI ID F和BsmBI ID R的PCR反应液中,进行PCR扩增和电泳检测。
1.2.6 puromysin 筛选浓度的测定 配置0、0.25、0.5、0.75、1、1.25 μg/mL 共6 个 浓 度 梯 度 的puromysin 溶液处理细胞48 h,拍照,根据细胞形态变化,确定puromysin细胞处理浓度。
1.2.7 质粒转染与有限稀释法建立OBSCN敲除突变的单细胞克隆 培养细胞融合度达50%左右时,使用无内毒素质粒reOBSCN和lipo 2000试剂转染,在转染之前使细胞在饥饿状态下培养,4 μL reOBSCN 和200 μL 空白培养基室温平衡5 min,含lipo 2000(混合比例为50∶1)的培养基室温平衡5 min,二者混合后平衡20 min,转染4 h后更换含血清正常培养基,培养6 h,更换含puromysin 的培养基,转染48 h后,更换含血清正常培养基,细胞成团生长后,采用有限稀释法进行单细胞克隆铺板,培养4 h后取出96孔板,标记出只有1个细胞的孔,第4天和第8天对单细胞孔进行换液处理,11~13天,对优势生长细胞株消化传代,扩大培养,得到敲除对照空载体HepG2 ecas 稳转细胞系和OBSCN敲除HepG2 OBSCN KO稳转细胞系。
1.2.8 Western blot蛋白免疫印迹实验 提取细胞蛋白,经BCA 定量后,电泳,湿转法将蛋白转移至PVDF 膜上,恒压法转膜,10%脱脂奶粉室温封闭1.5 h,一抗4℃ 56 r/min 在摇床上孵育12~16 h,TBST 洗液洗涤3 次,二抗室温孵育1~2 h,TBST 洗液洗涤3次,ECL发光液显影。
1.2.9 CCK-8 法检测细胞增殖能力 取处于对数生长期的细胞,以5 000个/孔的细胞浓度接种于96孔板上,每组重复3次,每24 h进行CCK-8检测。连续检测6 d。
1.2.10 Transwell 实验检测细胞迁移能力 取处于对数生长期的细胞,使用不含血清的DMEM培养基重悬细胞,取1 × 105/200 μL 接种于上室,并在下室加入500 μL 完全培养基,72 h 后取出小室,PBS洗涤3次后,用预冷甲醇固定半小时,棉签轻轻擦去上室细胞,每擦1 次用PBS 洗涤1 次,用DAPI 染液染色后清洗、拍照并计数。
1.3 统计学分析
分析数据均用均数 ± 标准差来表示,组间的比较采用student-t检验,统计分析以及绘图均采用GraphPad Prism 8.4.3 统计软件进行。检验水准为α= 0.05。
2 结 果
2.1 荧光定量PCR 验证HepG2 细胞有无OBSCN本底表达
采用荧光定量PCR 技术验证HepG2 细胞有无OBSCN本底表达,根据定量数值及分析(表3)、GAPDH 内参溶解曲线(图1A)和OBSCN目的基因溶解曲线(图1B),可以得知HepG2 细胞无OBSCN本底表达。
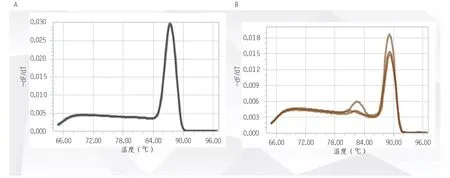
图1 荧光定量PCR验证HepG2细胞有无OBSCN本底表达
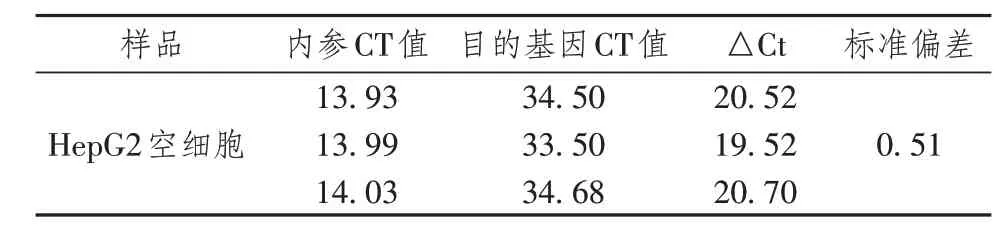
表3 HepG2细胞qPCR定量数值及分析
2.2 慢病毒滴度测定结果
针对病毒载体pSLenti-CMV-OBSCN(3 250 ~4 749 bp, G1333R)-3xFLAG-PGK-Puro-WPRE,进行慢病毒滴度测定,得到H21157 病毒原液滴度为5.66 × 108(IU/mL)。
2.3 慢病毒感染HepG2 细胞构建OBSCN 过表达细胞系
按照表4 所示病毒量感染细胞,慢病毒感染HepG2细胞72 h后,对HepG2(Blank)、HepG2 GL119、HepG2 H21157细胞拍照(图2A),可以看到慢病毒感染复数MOI为20时HepG2细胞状态良好;慢病毒感染HepG2 细 胞,puromysin 筛 选14 d 后,对HepG2 GL119、HepG2 H21157细胞拍照(图2B),可以看到使用puromysin筛选14 d后HepG2细胞状态良好。

图2 慢病毒感染HepG2细胞形态(× 100)

表4 不同滴度对应的病毒量
2.4 重组质粒reOBSCN的PCR鉴定结果
为验证重组质粒是否已经构建成功,利用普通PCR 扩增及琼脂糖电泳的方法验证扩增片段的大小是否与预期一致,质粒PCR琼脂糖电泳结果中泳道1~3分别为Marker、空载体对照质粒和reOBSCN重组质粒,可以看到泳道3 在308 bp 处有条带,2 000 bp 处无条带(图3A),证明reOBSCN 构建成功,菌落PCR 琼脂糖电泳结果中泳道1~3 分别为Marker、对照空载体PCR 反应液和reOBSCN 重组质粒菌落PCR 反应液,可以看到泳道3 也就是reOBSCN重组质粒菌落的PCR反应液在308 bp处有明显条带(图3B),进一步证明reOBSCN构建成功。

图3 琼脂糖电泳结果
2.5 重组质粒reOBSCN的测序结果
对上述已经验证条带大小正确的含重组质粒菌液测序,对所得到的正向序列测序结果(图4A)和反向序列测序结果(图4B)进行分析比较,进一步验证重组质粒reOBSCN是否构建成功,正向和反向序列测序结果均显示reOBSCN 与设计的引导RNA 引物序列(小写斜体加粗标记)一致,证明重组质粒reOBSCN构建成功。

图4 reOBSCN重组质粒双向测序结果
2.6 HepG2细胞饥饿状态及puromysin筛选浓度
为探究血清对HepG2细胞形态的影响,对细胞进行了24 h 内饥饿状态的探究(图5A),分别于0、4、8、10、20、24 h拍照,最终确定每次实验前对细胞进行4 h 饥饿状态处理,以消除血清对后续实验的干扰。为探究HepG2 细胞的puromysin 抗性浓度,对细胞进行了puromysin 抗性筛选(图5B),采用含0、0.25、0.5、0.75、1、1.25 μg/mL的puromysin培养液处理细胞48 h,拍照,根据细胞形态变化,最终确定0.75 μg/mL 用 做 后 续 实 验 中HepG2 细 胞 的puromysin处理浓度。
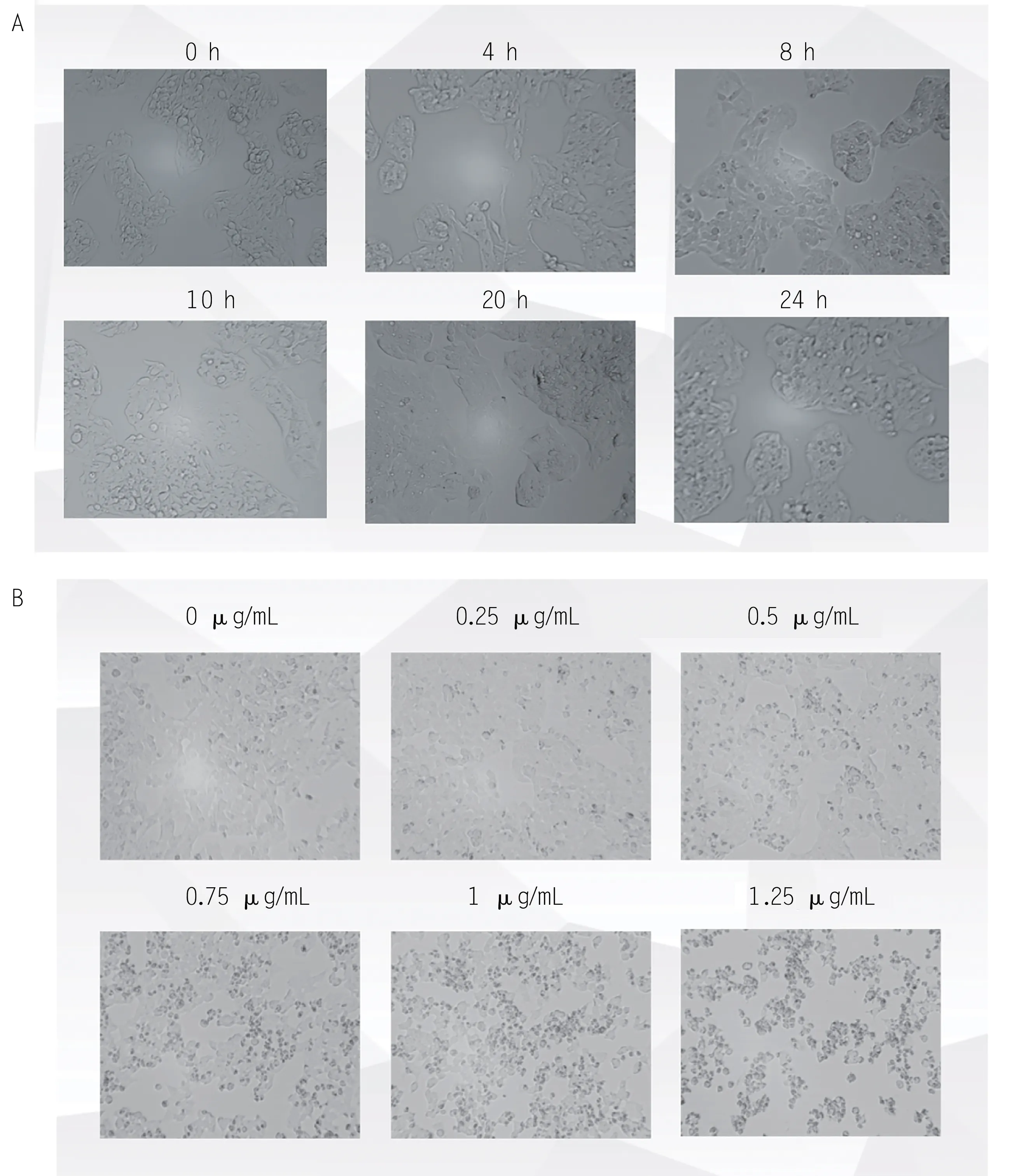
图5 HepG2细胞形态(× 100)
2.7 Western blot 蛋白免疫印迹实验检测Obscurin蛋白表达量
在蛋白表达水平上观察5株细胞Obscurin蛋白表达含量变化(图6)。构建的4株细胞系和HepG2细胞中目的蛋白Obscurin与内参蛋白Tubulin的比值,其中HepG2 细胞中Obscurin/Tubulin = 4.06,HepG2 H21157过表达细胞株中Obscurin/Tubulin = 6.98,相较于HepG2 GL119 过表达空载对照细胞株中Obscurin/Tubulin = 4.03,OBSCN蛋白表达水平明显升高,进一步证明OBSCN过表达载体构建成功;而HepG2 ecas 敲除空载对照细胞株中Obscurin/Tubulin = 6.69,相较于HepG2 OBSCN KO 敲除细胞株中Obscurin/Tubulin = 1.98,OBSCN蛋白表达水平降低,说明成功构建了OBSCN敲除表达载体。

图6 Western blot检测Obscurin蛋白表达量
2.8 CCK-8法检测细胞增殖能力
为探究OBSCN过表达及敲除细胞系对细胞增殖的影响,通过CCK-8法绘制细胞增殖曲线,第6天后细胞均到达平台期,故取前6 天的检测结果绘制细胞增殖曲线。第6 天可以明显看出HepG2 H21157 稳定表达细胞系相较于HepG2 GL119 稳定表达细胞系增殖加快(图7A),差异有统计学意义(P< 0.000 1),说明OBSCN过表达加快HepG2肝癌细胞的增殖;而HepG2 ecas 稳定表达细胞系与HepG2 OBSCN KO 稳定表达细胞系相比,HepG2 OBSCN KO 稳定表达细胞系增殖减慢(图7B),差异有统计学意义(P< 0.000 1),由此可以说明OBSCN的敲除会减慢HepG2肝癌细胞的增殖。

图7 CCK-8法检测HepG2细胞增殖能力
2.9 Transwell法检测细胞迁移能力
利用Transwell 小室法,检测OBSCN突变对HepG2肝癌细胞迁移能力的影响。HepG2 H21157与HepG2 GL119相比,72 h迁移细胞数见表5,可以看出HepG2 H21157 稳定表达的细胞系迁移细胞数为150.30 ± 14.95,HepG2 GL119 迁移细胞数为136.50± 15.02,可知OBSCN过表达使HepG2肝癌细胞迁移加快(图8 A);而HepG2 ecas 与HepG2 OBSCN KO 相比,72 h迁移细胞数见表5,HepG2 ecas迁移细胞量为147.80 ± 11.55,HepG2 OBSCN KO 迁移细胞量为112.70 ± 20.30,差异有统计学意义(P< 0.01),可知OBSCN的敲除会减慢HepG2 肝癌细胞的迁移(图8B)。
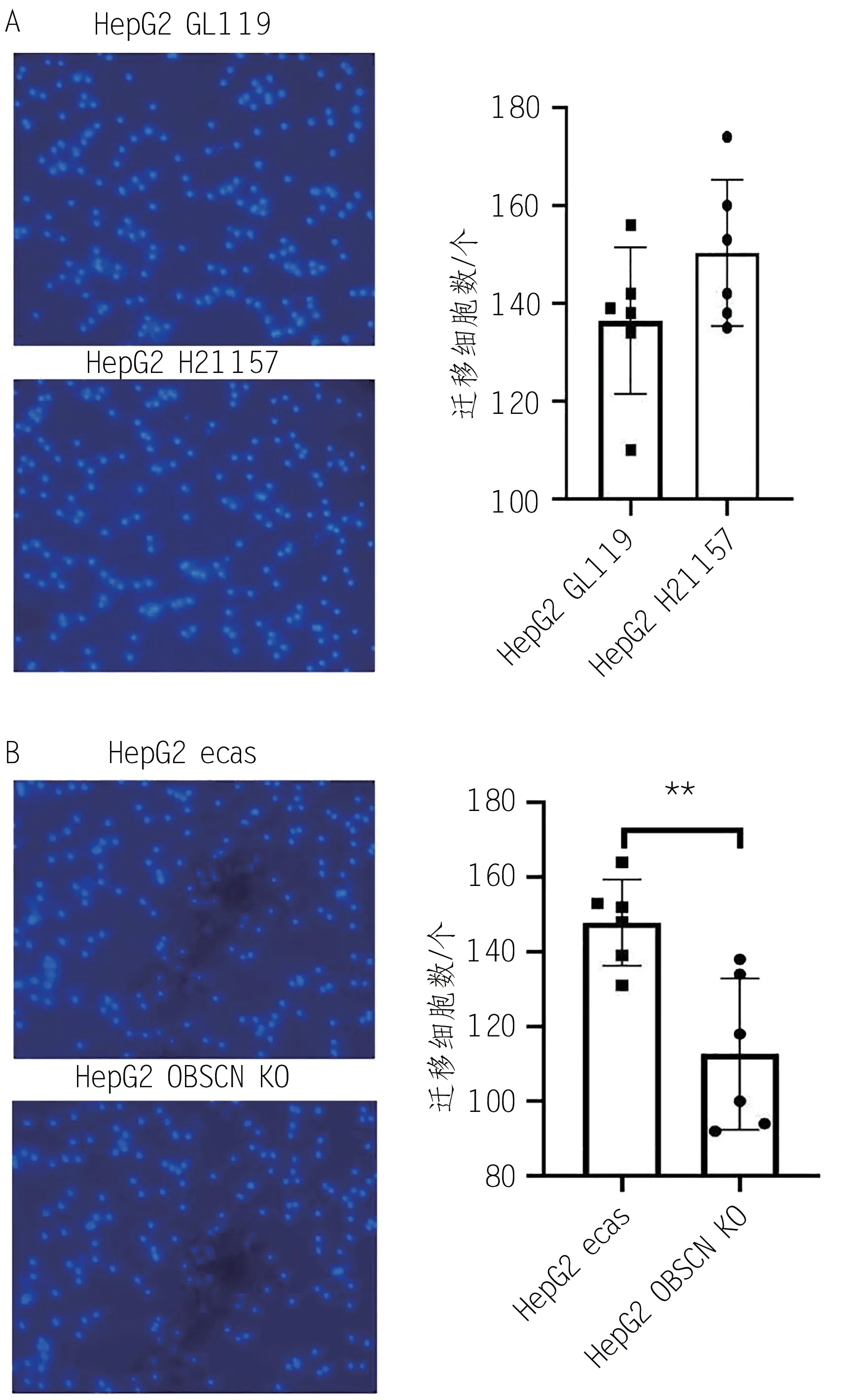
图8 Transwell法检测HepG2迁移能力(DAPI染色,× 200)

表5 不同细胞72 h迁移细胞数
3 讨 论
2020年全球癌症负担数据显示,因肝癌死亡的人超过83万[12]。但其发生发展机制尚不明确,总体预后不良。而细胞骨架蛋白OBSCN是体细胞突变频率较高的基因,位于lq42,跨越170 kb,并受到其119个外显子的选择性剪接,产生多种转录本,从而得到一个分子量跨度极大的蛋白质族群,基因序列长,功能区复杂[13]。
OBSCN主要存在于脊椎动物横纹肌组织中,在肌原纤维形成过程中发挥多种作用,包括提供整体结构完整性和肌原纤维稳定性,更具体地说是提供例如肌球蛋白等相关蛋白的适当空间定位[14]。有研究表明,通过对突变频率在肝癌样本中大于5%的TP53、TTN、CTNNB1、MUC16、PCLO、RYR2、OBSCN等45 个关键致病基因作为预后因素,进行生存分析和单因素cox 比例风险模型分析,单因素cox比例风险模型分析显示OBSCN与预后有显著相关性,且在生存分析中也被认为对预后有显著影响[15]。根据OBSCN在横纹肌和乳腺上皮细胞内发挥的支持骨架和信号传递的作用,推测其对维持肝癌细胞组织和促进信号转导有关键性作用,进而可以缓解肝癌患者的不良预后。
本研究利用荧光定量PCR 技术确定HepG2 肝癌细胞无OBSCN本底表达,利用慢病毒感染的方法,成功构建OBSCN过表达稳定表达细胞系,Crispr Cas9 技术构建OBSCN敲除重组质粒reOBSCN,随后经菌落PCR、质粒PCR 以及基因测序均验证其构建成功,将reOBSCN转染进HepG2细胞中,从而得到OBSCN敲除稳定表达细胞系,蛋白免疫印迹实验验证敲除及过表达细胞系构建成功。通过CCK-8法、transwell小室实验验证得知,OBSCN过表达会加快HepG2 肝癌细胞的增殖和迁移,而OBSCN基因的敲除则会减慢HepG2 肝癌细胞的增殖和迁移。
综上所述,OBSCN基因突变可以影响肝癌细胞的增殖和迁移,在肝癌的发生发展中起到了重要作用。本研究成功构建了OBSCN突变的稳定表达细胞系,为进一步深入探究OBSCN突变对肝癌细胞的生物学调控作用奠定了基础。研究以期寻找可行的药物作用靶点,从而为肝癌的不良预后现状提供理论支持。
利益冲突所有作者均声明不存在利益冲突
猜你喜欢
华人时刊(2022年9期)2022-09-06
华人时刊(2020年15期)2020-12-14
食品科学(2018年10期)2018-05-23
山东医药(2015年14期)2016-01-12
广州大学学报(自然科学版)(2015年4期)2015-12-23
西南医科大学学报(2015年1期)2015-08-22
江苏大学学报(医学版)(2015年2期)2015-04-17
中国当代医药(2015年9期)2015-03-01
中国医药导报(2015年26期)2015-02-28
西南军医(2015年6期)2015-01-23
