
盐酸度洛西汀工艺中类Hofmann-Martius重排产物的确证与产生机制
2023-09-11 08:33:54张可可林金生郑乐伟王吉超沈卫阳
中国药科大学学报 2023年4期
张可可,林金生,郑乐伟,王吉超,李 敏*,沈卫阳
(1浙江华海药业股份有限公司高等分析技术中心,临海 317024;2中国药科大学理学院,南京 211198)
抑郁症是一种常见且对人类身心健康有着严重危害的精神类疾病。盐酸度洛西汀(图1-A)作为一种新型的口服选择性5-羟色胺(5-HT)和去甲肾上腺素(NE)双重再摄取抑制剂(SNRI)在抑郁症的治疗中发挥了重要作用,主要用于治疗重度抑郁症、广泛性焦虑障碍、纤维肌痛和应激性尿失禁[1-2]。
在盐酸度洛西汀粗品与盐酸度洛西汀成品某一批次样品生产过程的质量控制中,发现了2 个未知单杂,其相对含量分别为0.27%与0.61%。为有效控制本品的内在质量,对以上两个未知单杂的结构、产生机制和盐酸度洛西汀的稳定性进行了相关考察,确证了以上两个未知杂质是盐酸度洛西汀在酸性条件下产生的两个类Hofmann-Martius重排产物,杂质1(图1-B)为萘酚环的对位重排产物,杂质2(图1-C)是萘酚环的邻位重排产物,其化学结构式分别见图1。因二者的产生与酸性条件有关,故随后考察了结晶过程合适的pH和温度,结果显示在50 ℃以下、pH 3 ~ 7 的条件下盐酸度洛西汀几乎不会发生分解。结合稳定性考察结果及实际的生产工艺条件,选择丙酮作为结晶溶剂进行了重结晶实验,结果显示得到盐酸度洛西汀成品的纯度显著提升,且收率达到了85%以上。

Figure 1 Structure of duloxetine hydrochloride (A), para-rearrangement product (B) and ortho-rearrangement product (C)
1 仪器与试药
戴安Ultimate 3000高效液相色谱仪(美国赛默飞世尔公司);Agilent LC-MS 6120液质联用仪、Agilent 1260&6545 Q-TOF 四极杆飞行时间液质联用仪、400-MR DD2 核磁共振波谱仪(美国安捷伦科技有限公司)。
盐酸度洛西汀原料药(批号:JSW-1051-31-01、2935-19-008、SYP-1595-05-03、20081-23-094PV,浙江华海药业股份有限公司);甲醇[色谱纯,批号:WXBD5460V,西格玛奥德里奇(上海)贸易有限公司];乙腈[色谱纯,批号:21035178,霍尼韦尔贸易(上海)有限公司];二甲基亚砜(色谱纯,批号:21B4345)、N,N′-二甲基甲酰胺(色谱纯,批号:20E3588)、二氯甲烷( 色谱纯,批号:K53445844124)、N-甲基吡咯烷酮(色谱纯,批号:K51223597912) (美国默克公司);异丙醇(色谱纯,批号:21C6220)(美国ROE公司);其他试剂均为市售分析纯。
2 实验条件
两个重排产物是在不同批次的盐酸度洛西汀生产过程中发现的,为了保证两个产物各自的响应和分离度,在对以上两个重排产物进行质谱分析时,采用了不同的分析方法。
2.1 杂质1分析方法
2.1.1 色谱条件 色谱柱为Eclipse XDB-C8色谱柱(4.6 mm × 150 mm, 3.5 µm);柱温为40 ℃;检测波长为230 nm;流速为1 mL/min;进样量为10 µL;流动相:混合液(10 mmol/L 乙酸铵水溶液-乙腈,85∶15) (A)-乙腈(B),洗脱程序如下:0 ~ 5 min, 0%B; 5 ~ 20 min, 0% ~ 80% B; 20 ~ 25 min, 80% B;25 ~ 25.1 min, 80% ~ 0% B; 25.1 ~ 30 min, 0% B。
2.1.2 质谱条件 在ESI 源正离子模式下进行检测,扫描范围m/z100 ~ 400。干燥气流速为10 L/min,鞘气流速为12 L/min;干燥气温度为320 ℃,鞘气温度为350 ℃;Fragmentor 电压为60 V,喷嘴电压为600 V,毛细管电压为3 500 V;雾化气压力为60 psi (1 psi = 6.895 kPa);采集带宽为4 amu;采集速率为1 spectrum/s;二级碰撞能量为30/40/50 eV。
2.1.3 供试品溶液配制 取样品3 mg,精密称定置于10 mL 量瓶中,加入溶剂(甲醇-水,75∶25)溶解后稀释至刻度。
2.1.4 酸降解条件 取样品5 g,溶解于乙酸乙酯100 mL,加入浓盐酸2 mL,于40 ℃条件下反应45 min,反应完成后,将反应液旋干,稀释后进样检测。
2.1.5 核磁共振检测条件 核磁共振信息在Varian 400 MHz核磁共振波谱仪上测定,质子频率为400 MHz,实验的温度控制在23 ℃,溶剂氘代为二甲基亚砜。
2.2 杂质2分析方法
2.2.1 色谱条件 色谱柱为Eclipse XDB-C8色谱柱(4.6 mm × 150 mm, 3.5 µm);柱温:40 ℃,检测波长230 nm;流速为1.0 mL/min;进样量为10 µL;流动相:缓冲液(氨水0.2 mL 加入水1 000 mL 用三氟乙酸调pH至3.0)-乙腈(70∶30),运行时间为20 min。
2.2.2 质谱条件 在ESI 源正离子模式下进行检测,扫描范围m/z50 ~ 1 700。干燥气流速为12 L/min,鞘气流速为12 L/min;干燥气温度为300 ℃,鞘气温度为320 ℃;Fragmentor电压为70 V,喷嘴电压为600 V,毛细管电压为3 500 V;雾化气压力为60 psi;采集带宽为4 amu;采集速率为1 spectrum/s;二级碰撞能量为30/40/50 eV。
2.2.3 供试品溶液配制 取20 mg 样品,精密称定置于5 mL 量瓶中,加入溶剂(乙腈-水,75∶25)溶解后稀释至刻度。
2.2.4 核磁共振检测条件 核磁共振信息在核磁共振波谱仪上测定,质子频率为400 MHz,实验的温度控制在23 ℃,溶剂为氘代氯仿。
2.3 两个类Hofmann-Martius 重排产物的稳定性考察试验
2.3.1 加热条件及不同溶剂条件中产物分布考察
(1)条件1:60 ℃取样品20 mg,精密称定置于20 mL 顶空瓶,平行称取9 份,分别加入水、甲醇、乙腈、丙酮、乙酸乙酯、二甲基亚砜、N,N′-二甲基甲酰胺,N-甲基吡咯烷酮和异丙醇10 mL,压盖密封,混匀后置于60 ℃条件下加热反应,于15 h 和24 h分别取样检测。
(2)条件2:80 ℃取样品20 mg,精密称定置于20 mL 顶空瓶,平行称取9 份,分别加入水、甲醇、乙腈、丙酮、乙酸乙酯、二甲基亚砜、N,N′-二甲基甲酰胺,N-甲基吡咯烷酮和异丙醇10 mL,压盖密封,混匀后置于80 ℃条件下加热反应,于5、8、15、20和24 h分别取样检测。
2.3.2 白光光照条件下产物分布考察 取样品30 mg,精密称定置于透明塑封袋,平行称取9 份,塑封,置于平均照度为8 390.4 lx 的白光光照条件下,分别于1、2、3、5、7、10、15、18、25 d 时取出,置于25 mL 量瓶中,用溶剂(乙腈-水,75∶25)溶解后稀释至刻度,后取出250 µL 加入溶剂750 µL 稀释为0.3 g/L。
2.3.3 紫外光照条件下产物分布考察
(1)盐酸度洛西汀固体原料药:取样品30 mg,精密称定置于透明塑封袋,平行称取3份,塑封,置于平均照度为1 377.4 mW/cm2的紫外光照条件下,分别于1、2、3 d 时取出,置于25 mL 量瓶中,用溶剂(乙腈-水,75∶25)溶解后稀释至刻度,后取出250 µL加入溶剂750 µL稀释为0.3 g/L。
(2)盐酸度洛西汀溶液:取样品30 mg,精密称定置于比色皿中,加入溶剂(乙腈-水,75∶25) 10 mL 溶解,置于平均照度值为1 377.4 mW/cm2的紫外光照条件下反应,于1、2、3 d 时分别取出100 µL加入溶剂900 µL稀释为0.3 g/L。
2.3.4 高温固态条件下产物分布考察 取样品300 mg,精密称定置于20 mL 称量瓶中,平行称取6份,置于80 ℃条件下,分别于1、2、3、8、10、15 d时取出,置于100 mL 量瓶中,用溶剂(乙腈-水,75∶25)溶解后稀释至刻度,后取出100 µL 加入溶剂900 µL稀释为0.3 g/L。
2.3.5 高温高湿固态条件下产物分布考察 取样品300 mg,精密称定置于20 mL 称量瓶中,平行称取6 份,置于80 ℃、75% RH 条件下,分别于1、2、3、8、10、15 d 时取出,置于100 mL 量瓶中,用溶剂(乙腈-水,75∶25)溶解后稀释至刻度,后取出100 µL加入溶剂900 µL稀释为0.3 g/L。
2.4 pH和温度考察
取适量丙酮溶液4 份,分别调pH 至1.2、2.5、3.5、7,备用。
取样品20 mg,精密称定置于20 mL顶空瓶中,平行称取4 份,分别用pH 为1.2、2.5、3.5、7 的丙酮溶液稀释至2 mg/mL,置于室温下反应6 h。
取样品20 mg,精密称定置于20 mL顶空瓶中,平行称取4 份,分别用pH 为1.2、2.5、3.5、7 的丙酮溶液稀释至2 mg/mL,置于40 ℃下反应6 h。
取样品20 mg,精密称定置于20 mL顶空瓶中,平行称取4 份,分别用pH 为1.2、2.5、3.5、7 的丙酮溶液稀释至2 mg/mL,置于50 ℃下反应6 h。
取样品20 mg,精密称定置于20 mL顶空瓶中,平行称取4 份,分别用pH 为1.2、2.5、3.5、7 的丙酮溶液稀释至2 mg/mL,置于60 ℃下反应6 h。
2.5 重结晶实验
取盐酸度洛西汀粗品2 g,精密称定于250 mL圆底烧瓶中,加入丙酮200 mL,加热使其完全溶解,2 h 内缓慢冷却至室温,接着继续冷却至5 ℃左右,继续析晶1 h,过滤,滤饼用冷却至5 ℃的丙酮10 mL洗涤,烘干。
3 结果与讨论
3.1 杂质1(对位重排产物)及杂质2(邻位重排产物)定位分析
采用原始色谱分析方法采集的盐酸度洛西汀样品的色谱图(图2-A)中,度洛西汀在15.8 min 出峰,在6.5min 处出现一个未知单杂,含量为0.27%,为了表述方便,将该杂质命名为杂质1。采用原始色谱分析方法采集的某一批次的盐酸度洛西汀样品的色谱图(图2-B)中,度洛西汀在16.3 min 出峰,在11.9 min 处出现一个未知单杂,含量为0.61%,为了表述方便,将该杂质命名为杂质2。由于原始色谱分析方法不适用于质谱检测器,并且为了保证这两个未知杂质各自的响应和分离度,针对两个批次的样品分别开发了新的液质联用分析方法,即实验条件中的杂质1 分析方法和杂质2分析方法。

Figure 2 UV chromatogram of duloxetine hydrochloride
3.2 杂质1(对位重排产物)结构解析
采用液质联用分析方法对盐酸度洛西汀样品分析时,通过采集该杂质的质谱信息,得知其[M+H]+的精确质荷比为m/z298.122 7,匹配分子式为C18H20NOS,匹配误差为11 × 10-6,分子式与度洛西汀相同,由此可确定,该杂质为度洛西汀的同分异构体。
由于杂质1于度洛西汀成盐过程中生成,在该步成盐工序中,有盐酸的加入,同时度洛西汀分子中噻吩环苄位含有一个萘酚醚基团,在酸性条件下容易发生醚键断裂并重排,因此怀疑杂质1的生成与酸有关,于是进行了相关的酸降解实验。通过酸降解,在相同的保留时间产生了大量降解产物,含量为29.1%,该降解产物的[M+H]+的精确质荷比为m/z298.122 7,匹配分子式为C18H20NOS,误差11 × 10-6。先后采集了该降解物与目标杂质的紫外吸收光谱图和二级质谱数据,结果显示完全一致,具体数据见表1。综上可以确认度洛西汀酸降解的目标降解物与盐酸度洛西汀粗品中的杂质1为同一化合物。
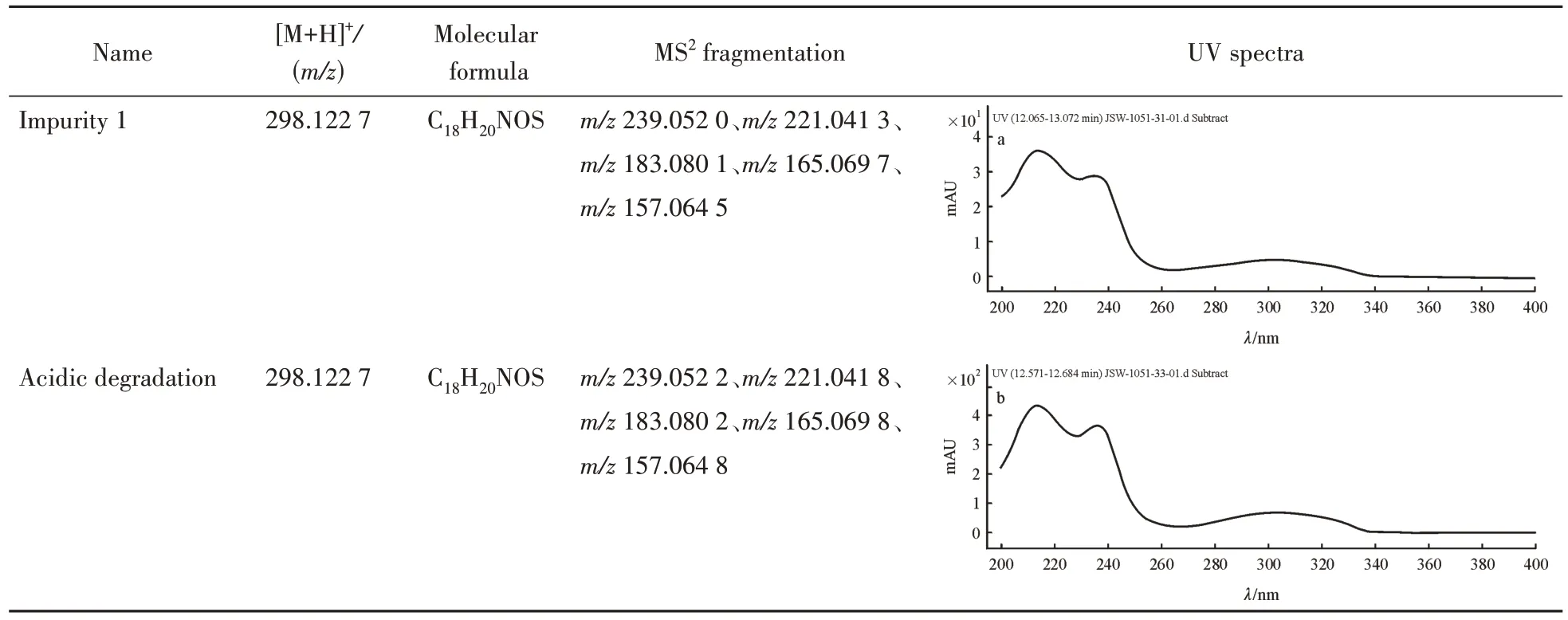
Table 1 Comparison of LC-UV-MS data of duloxetine and impurity 1
随后分离了该杂质,并对其进行了核磁共振检测,1H NMR和13C NMR谱归属的碳氢信号数据见表2。对照专利中的相关记载[3],确定该杂质为度洛西汀酸降解后的类Hofmann-Martius 对位重排产物。同时,对此杂质1的二级碎片及度洛西汀的二级碎片解析,也能支持目标杂质为度洛西汀的类Hofmann-Martius 对位重排物的结论。度洛西汀可能的裂解途径见图3,杂质1可能的裂解途径见图4。
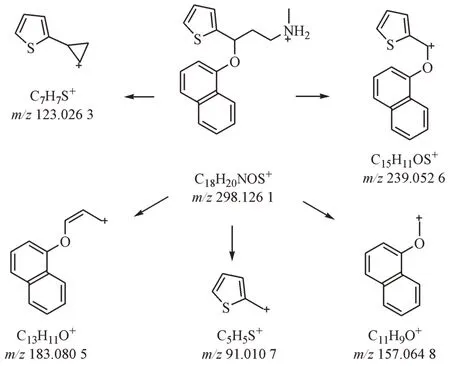
Figure 3 Proposed mechanism for MS2 fragmentation pathway of duloxetine
3.3 杂质2(邻位重排产物)结构解析
对盐酸度洛西汀样品在原始色谱分析方法中于11.9 min 出峰的杂质进行收集,即杂质2,色谱图见图2-B。随后将收集到的杂质2采用液质联用分析方法进行检测,通过采集该杂质的质谱信息,得知其[M+H]+的精确质荷比为m/z298.127 0,匹配分子式为C18H20NOS,匹配误差为3.4 × 10-6,其分子式与度洛西汀及杂质1均相同,但三者色谱出峰时间差异较大,由此可确定,该杂质为度洛西汀的另一同分异构体。
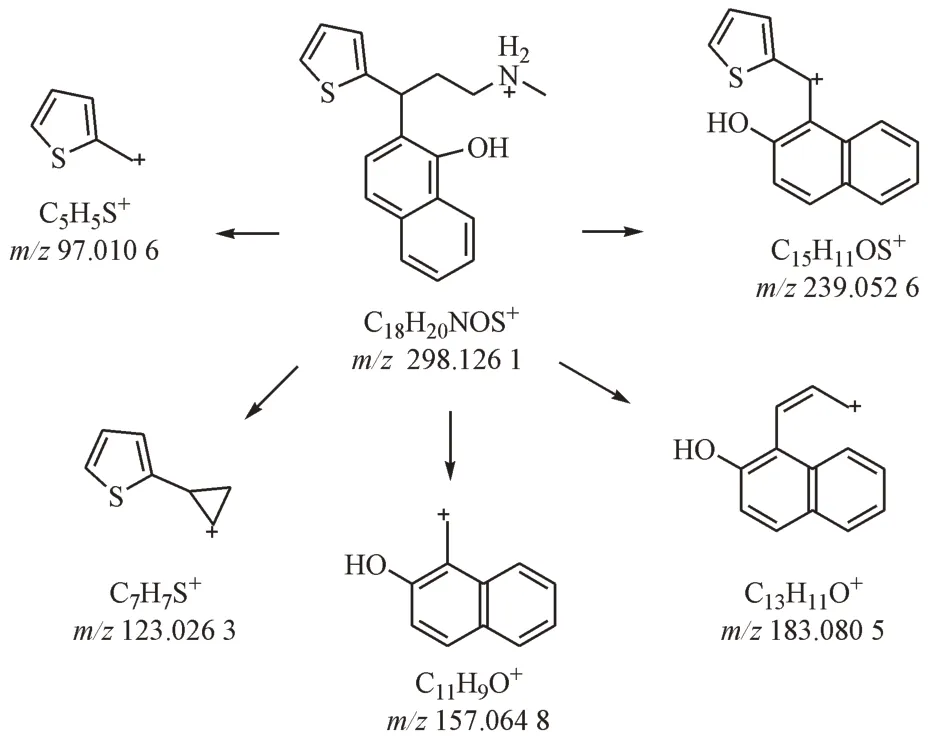
Figure 4 Proposed mechanism for MS2 fragmentation pathway of impurity 1
首先将该杂质与度洛西汀的紫外吸收光谱图进行了对比,见图5,发现该杂质与度洛西汀的紫外吸收较为相似,二者最大吸收波长分别都在215 nm和235 nm 左右,并且都在290 nm 左右有一个弱吸收。接着将该杂质和度洛西汀的二级质谱图进行对比,发现二者含有大量相同的碎片离子,如m/z239.052 5,m/z221.042 7,m/z183.080 9,m/z157.065 2。结合杂质1 的产生原因分析,推测盐酸度洛西汀在酸性条件下除了会产生其萘酚环对位重排产物外,还会产生萘酚环邻位重排产物,即在萘酚中间态与烷基噻吩正离子发生亲核结合时,除发生对位重排反应外,应该还会发生邻位重排。结合杂质2 的结构解析推测杂质2 可能为度洛西汀的邻位重排产物。
为了确定该杂质的结构,分离并制备了该杂质,对其进行了核磁共振检测,1H NMR 和13C NMR谱归属的碳氢信号数据见表2。通过对1H NMR 谱和13C NMR 谱进行分析,可以确定杂质2 即是度洛西汀的类Hofmann-Martius邻位重排产物,为杂质1的同分异构体。杂质2可能的裂解途径见图6。
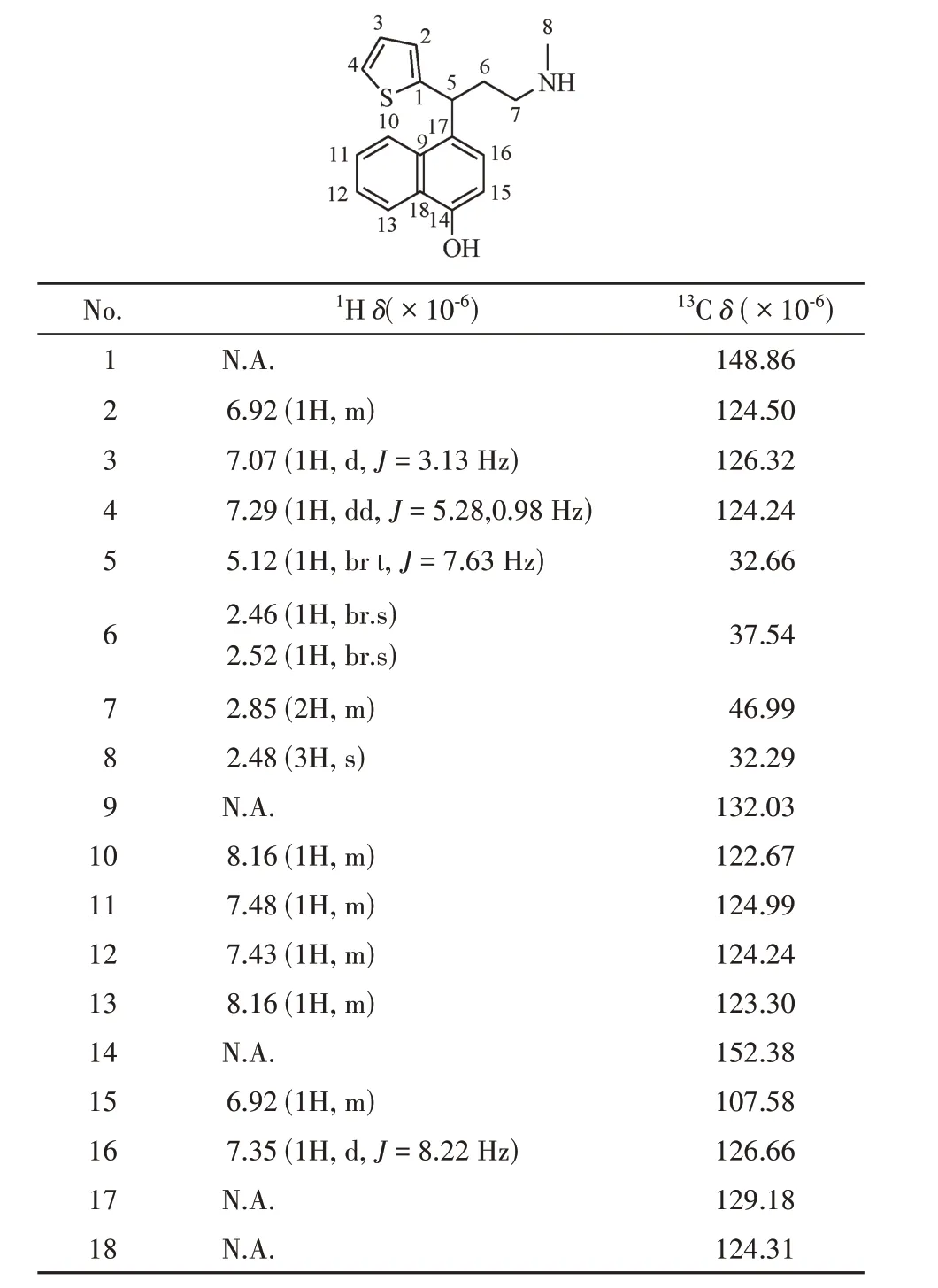
Table 2 1H NMR and 13C NMR data of the para-rearrangement product
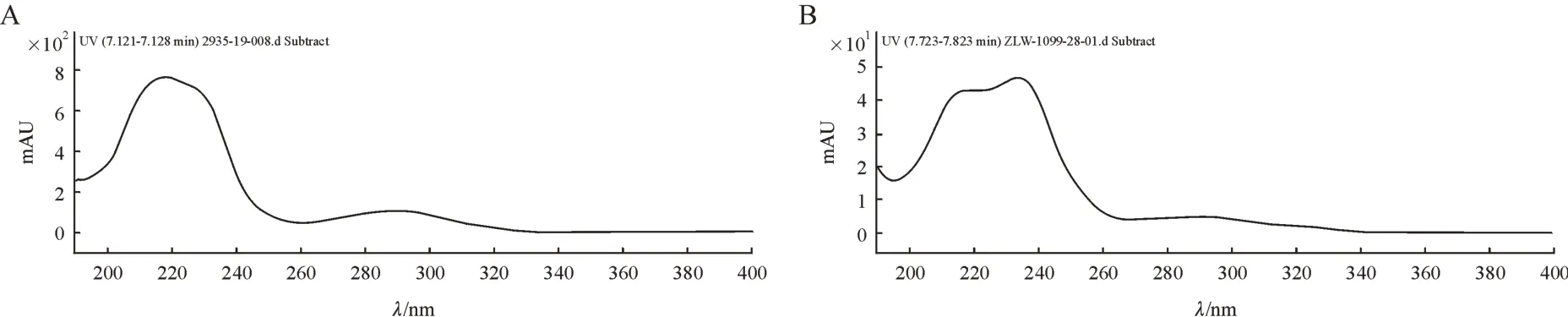
Figure 5 Comparison of UV spectra between duloxetine (A) and impurity 2 (B)
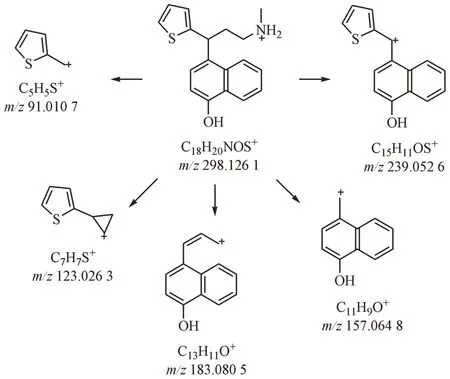
Figure 6 Proposed mechanism for MS2 fragmentation pathway of impurity 2
通过1H NMR、13C NMR谱分别对杂质1和杂质2进行解析,具体解析过程如下:通过对13C NMR谱进行分析,发现这两个化合物与度洛西汀相同,均含有18 个碳原子,包括1 个甲基碳信号,2 个亚甲基碳信号,1 个烷基次甲基碳信号,5 个季碳信号,同时含有9个芳基次甲基碳信号,但这两个化合物5-位上的碳原子信号相比度洛西汀明显向高场移动,说明5-位上的碳原子可能已经不再和氧原子相连,具体的13C 谱信号见表2。1H NMR 谱中显示化合物含有与相应碳原子对应的17 个质子信号,通过其化学位移、积分,确定这两个化合物均含有3 个甲基氢原子,2 组亚甲基氢原子(共4 个氢原子)、1个烷基次甲基氢原子以及9个芳基次甲基氢原子信号,这与13C谱信号相符,同时发现羟基和氨基位置的活泼氢没有出峰。同时结合文献数据[3],确定杂质1 和杂质2 分别为度洛西汀的对位和邻位的取代产物。
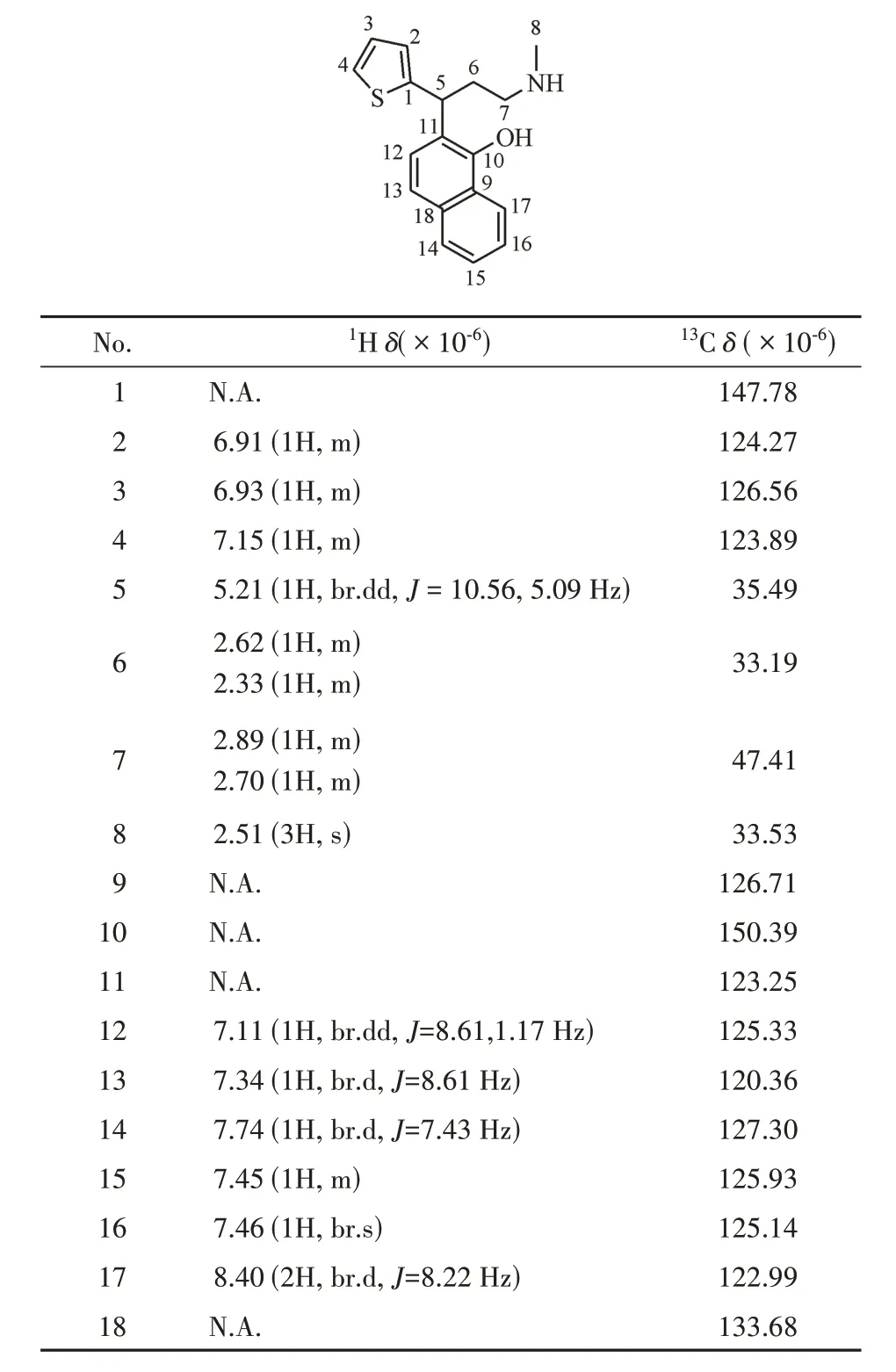
Table 3 1H NMR and 13C NMR data of the ortho-rearrangement product
3.4 两种类Hofmann-Martius 重排产物的稳定性考察结果
经过以上条件的考察,两个重排产物主要的产生情况如下:盐酸度洛西汀80 ℃条件下以水为溶剂反应至5 h 时,就产生了两种类Hofmann-Martius 重排产物,对位和邻位重排产物的含量分别为2.21%和11.03%;反应至8 h 时产生的两种重排产物的含量分别为3.75%和20.23%;至15 h 时,度洛西汀已出现降解过度现象,二者的含量分别为8.82%和39.01%。以甲醇为溶剂的条件下反应至8 h 时产生的对位和邻位重排产物的含量分别为0.44%和0.24%;反应至24 h 时,二者的含量分别达到了1.49%和0.79%。以乙腈为溶剂反应至8 h 时产生了含量为0.16%的对位重排产物;反应至24 h 时,二者的含量分别为0.61%和0.26%。以异丙醇为溶剂时,反应至24 h 时,两种重排产物的含量分别为0.71%和0.11%。两种重排产物在以水、甲醇、乙腈和异丙醇为溶剂在60 ℃条件下的含量随时间的变化趋势分别见图7-A 和图7-B;在80 ℃条件下的含量随时间的变化趋势分别见图7-C和图7-D;在60 ℃和80 ℃条件下以其他试剂作溶剂时,二者均没有产生;盐酸度洛西汀溶液在紫外灯下反应至第3 天时有少量对位重排产物产生;盐酸度洛西汀固体原料药在高温高湿(80 ℃、75% RH)条件下反应至第10天时有少量的对位重排产物的产生;在其他的考察条件下均没有目标杂质的产生。
对以上考察结果进行分析,两种重排产物的产生可能与水等质子性溶剂有关,与溶剂的质子化程度呈正相关,且二者的产量随温度的升高而增大,如在现有的考察条件下,在80 ℃以水为溶剂时,两种重排产物的含量最大。这一结论正好与两种重排产物的产生条件与酸有关的推测相契合,盐酸度洛西汀的盐酸部分在质子性溶剂中会以游离态的形式存在,为重排反应的发生提供了必要的酸性环境,温度升高则可以加剧反应的进行。同时,对非质子性溶剂也进行了考察,发现其中乙腈虽然为非质子性溶剂,但是有文献报道,乙腈中含有少量的氢氰酸存在[4-5],故乙腈中也存在一定的酸性条件,这可能是两种类Hofmann-Martius重排产物在乙腈中也有少量生成的原因。盐酸度洛西汀固体原料药在高温高湿(80 ℃、75% RH)条件下有少量的对位重排物的产生,提示药物稳定性研究过程中,对位重排物杂质可能会增长,需要关注在存储过程中该杂质是否有超标的风险。
3.5 两个类Hofmann-Martius 重排产物可能的产生机制
对于以上两个类Hofmann-Martius 重排产物,Arava 等[6-7]发表过的关于度洛西汀在酸性条件下发生重排反应的报道,该报道显示,度洛西汀在某些条件下可以发生类Hofmann-Martius 重排反应,生成以对位取代产物为主并且有少量邻位取代产物产生的萘酚类重排产物,但在论文中只提到此两个重排产物是在某些极端的破坏性条件,比如在特定溶剂及Lewis 酸三氯化铝或三氟化硼存在时可以产生,但这两个试剂在实际工艺中并未使用到,与实际工艺条件下重排产物的产生情况有很大的不同,并且此论文也未对杂质的产生机制进行研究,因此研究工艺条件下重排产物产生条件及其产生机制研究具有重要的现实意义。
在本论文的研究中,先是对实际生产过程中遇到的两个重排产物的结构进行了解析,后又考察了在不同温度及不同溶剂中、相关固态降解条件下两个重排产物的分布情况,以上研究对盐酸度洛西汀的实际生产具有重要参考意义,同时,也对此两个重排物的产生机制进行了深入研究。实验结果表明,两个重排产物的产生与酸性环境密切相关,在酸性条件下,度洛西汀(1)中萘酚醚基团的醚氧键发生断裂,接着产生的萘酚中间态(2)和烷基噻吩正离子中间态(3),由于产生的烷基噻吩正离子中间态(3)具有很强的亲电性,将分别在萘酚环的邻位和对位的对萘酚中间体(2)发生亲核进攻产生两个同分异构体(4和5)。度洛西汀对位重排产物和邻位重排产物的产生机制分析见图8。
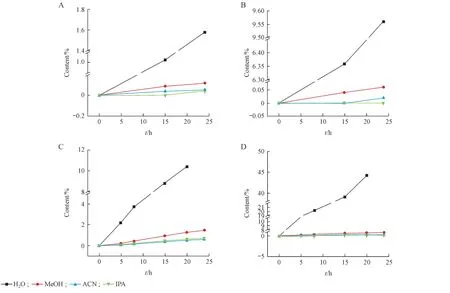
Figure 7 Level of para-rearrangement product and ortho-rearrangement product in each solvent with time at different temperature A: Level of pararearrangement product in each solvent with time at 60 °C; B: Level of ortho-rearrangement product in each solvent with time at 60 °C; C: Level of pararearrangement product in each solvent with time at 80 °C; D: Level of ortho-rearrangement product in each solvent with time at 80 °C
3.6 pH范围和温度考察结果
当使用丙酮当溶剂时,在常温、40 ℃、50 ℃条件下,同时pH 为2.5、3.5、7时,盐酸度洛西汀分子非常稳定,无降解杂质产生;而当pH 为1.2 时,在40 ℃、50 ℃条件下,可以发现盐酸度洛西汀均会发生降解产生大于0.5%的重排杂质1 及重排杂质2,同时也会产生大量的其他杂质。因此,在丙酮当结晶溶剂的条件下,pH 大于2.5 可以保证盐酸度洛西汀不会发生分解。同时,考虑到实际结晶工艺中过量的酸存在条件下,会导致最终成品的酸残留,并且盐酸度洛西汀溶解于丙酮本身就呈弱酸性,因此可以确认,采用丙酮当结晶溶剂对盐酸度洛西汀直接进行重结晶,既可保证杂质去除效果,又能有效避免盐酸度洛西汀降解产生这两个重排杂质。
3.7 重结晶实验结果
根据“2.5”项部分对盐酸度洛西汀粗品进行了重结晶研究,根据前期的实验结果,发现用丙酮当溶剂,且pH 在2.5 ~ 7 之间,盐酸度洛西汀的稳定性非常高,几乎完全不会降解产生这两个重排杂质及其他杂质。在重结晶实验中,起始物料盐酸度洛西汀粗品的纯度为99.0%,重排杂质1含量为0.25%,重排杂质2的含量为0.20%,重结晶之后样品的纯度为99.6%,重排杂质1 及重排杂质2 的含量均低于0.05%,收率为87%,结合两个重排产物的溶剂考察结果、重结晶实验结果以及生产工艺的可行性等因素,最终选择了非质子性溶剂丙酮作为了实际生产工艺的结晶溶剂。
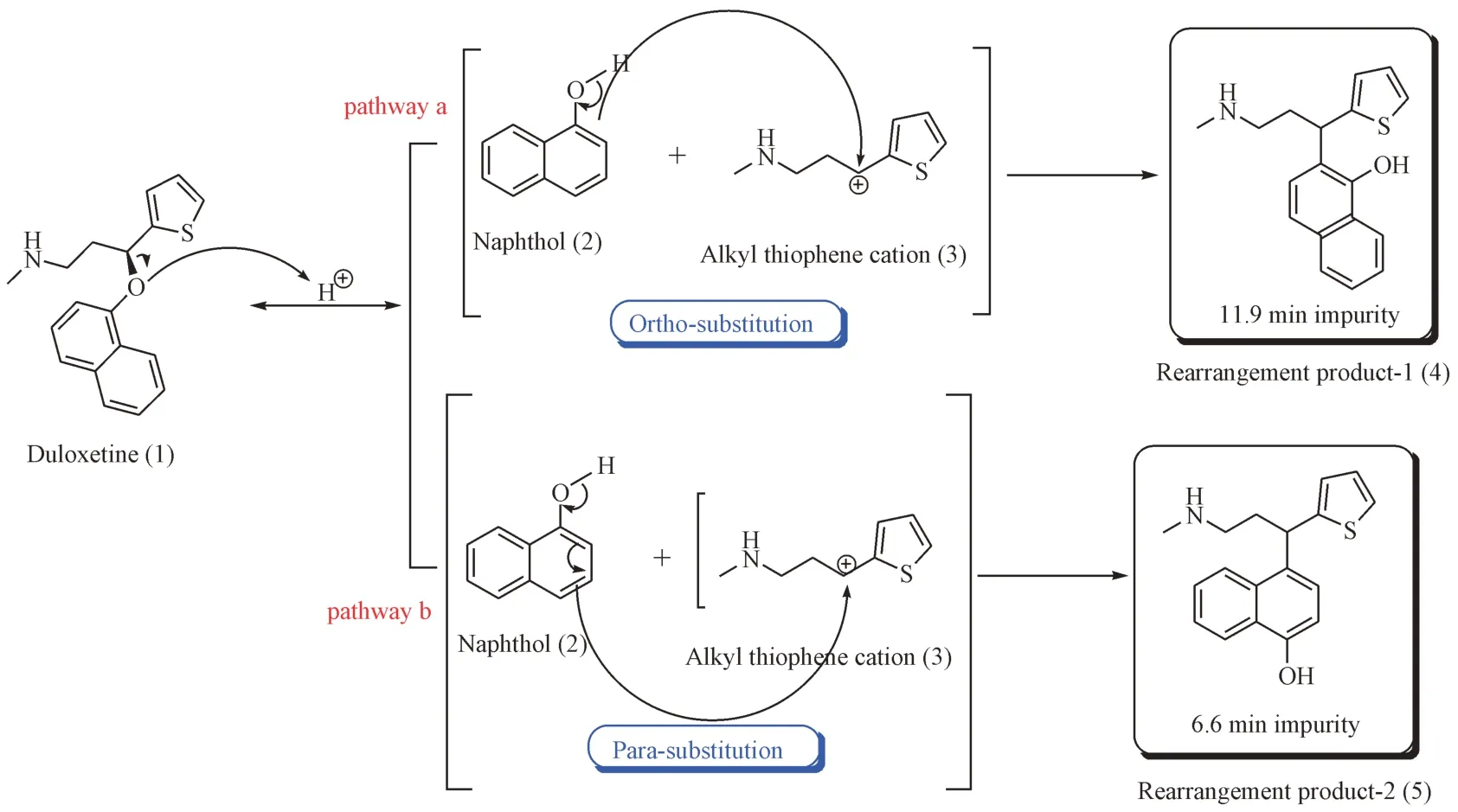
Figure 8 Proposed pathway for the formation of the para-rearrangement product (impurity 1) and the ortho-rearrangement product (impurity 2) in duloxetine hydrochloride
4 总 结
本研究对盐酸度洛西汀生产过程中发现的杂质1 和杂质2 进行了结构解析,结果证实两个杂质分别为盐酸度洛西汀成盐过程中,萘酚醚键断裂,并在萘酚环对位和邻位发生重排产生的同分异构体;同时对两个重排产物的稳定性和产生条件进行了考察,结果表明两个重排产物的产生与酸性条件及质子性溶剂的存在密切相关,并且这两个重排产物的产生受温度影响也非常大。在生产过程中这两个重排产物的发现,提示在盐酸度洛西汀生产工艺中应重点关注度洛西汀成盐过程中的pH 和温度变化,最终筛选出了有效的结晶溶剂丙酮,并将反应pH 控制在3 ~ 7,反应温度控制在50 ℃以下,以将两个重排产物的含量控制在相关指标之下,从而保证盐酸度洛西汀成品的质量。
猜你喜欢
天津音乐学院学报(2022年2期)2022-07-28 02:32:12
右江医学(2022年3期)2022-04-17 11:42:53
大学化学(2021年7期)2021-08-29 12:21:30
通信技术(2019年8期)2019-09-03 08:57:08
国际呼吸杂志(2019年4期)2019-03-12 01:08:08
印制电路信息(2015年6期)2015-12-30 12:57:52
四川精神卫生(2015年4期)2015-12-23 10:41:42
当代音乐(下旬刊)(2015年5期)2015-05-30 05:21:05
浙江理工大学学报(自然科学版)(2015年7期)2015-03-01 02:54:28
中国药业(2014年17期)2014-05-26 09:07:56
