
高通量分析人类粪便、皮肤和水环境中共享抗生素抗性基因的分布
2023-08-15 08:30:08周振超郑吉帅馨怡林泽俊陈红
生物技术通报 2023年7期
周振超 郑吉 帅馨怡 林泽俊 陈红
(1.浙江大学环境与资源学院环境技术研究所,杭州 310058; 2.宁波市生态环境科学研究院 宁波 315012)
抗生素抗性基因(antibiotic resistance genes,ARGs)被认为是一种新污染物已经在环境中被广泛发现[1]。研究已经在城市饮用水[2-4]、污水处理系统[5-7]和地表水[8-10]都中发现了丰富的ARGs,并且在人类微生物组中也发现了丰富的ARGs分布,例如肠道和皮肤微生物组中[11-14]。一部分ARGs可以被饮用水处理系统和污水处理系统等去除[3-5,7,15],甚至在环境中自然降解[8,16],但是存在一些ARGs难以被处理系统完全去除并且可以在环境中持续传播[17-19]。在不同环境中持续被检出的ARGs被认为是共享ARGs(shared ARGs)[20-22]。共享ARGs在不同环境样品中的丰度较高且能持续传播扩散,研究报道了在116个污水样本中共检测出381个不同的ARGs,其中发现了31个共享ARGs占检测到的总ARGs丰度的57.7%(范围29.4%-84.3%)[20]。已有研究也报道了河流生态系统中大约43.3%的ARGs在4个季节之间共享[21],而且水环境和鱼体中也存在共享ARGs[22]。ARGs可以通过细菌、可移动遗传元件(mobile genetic elements,MGEs)甚至游离态DNA进行转移和传播[23]。已有研究发现ARGs(例如macA-macB和tetA-tetR)在鸡、猪和人类粪便中共享[24],环境与人体中的共享ARGs可能会存在较大的传播和健康风险。环境和人类之间共享ARGs是否会影响临床抗生素耐药,共享ARGs是否仅限于特定机制的基因或适用于具有不同抗性机制的基因仍不清楚。因此,进一步探究环境中持续传播和共享ARGs分布和潜在宿主,可以为识别对公共健康构成较大威胁的ARGs和微生物群提供科学依据。
城郊区域存在着丰富的生物化学物质和元素交流及循环,也被认为是ARGs传播和扩散的热点区域之一[25]。团队前期研究报道了在城郊流域样品中检测到了46-154种不同的ARGs,且ARGs丰度受到季节和人为活动等因素的影响[16],且城郊居民粪便、皮肤和生活污水处理系统中的ARGs总丰度比河流样本中的丰度高约23、2和7倍[26]。但是,对城郊自来水、生活污水处理系统(RDSTS)、河流以及人体粪便和皮肤中的共享ARGs的分布和传播特征知之甚少。本研究采用高通量荧光定量PCR(highthroughput quantitative polymerase chain reaction, HT-qPCR)和16S rRNA 基因测序探究鉴定城郊区域水环境(自来水、污水和地表水)和人体(肠道和皮肤)中的共享ARGs和细菌标志物,探究ARGs的潜在宿主。这项研究有助于揭示城郊区域中关键的共享ARGs和微生物群落,以期为控制抗生素耐药传播提供理论依据。
1 材料与方法
1.1 材料
所有样本均在中国浙江省宁波市的一个城郊区域采集,连续3 d收集区域内的自来水(3个采样位点)、污水(2个采样位点)和地表水(2个采样位点),每个样品重复3次。在获得浙江大学医学院伦理委员会许可后,招募区域内10名健康成人(6男4女),采集受试者粪便和皮肤微生物样本。所有受试者在参与前都提供了书面知情同意书。使用无菌粪便收集器收集粪便样本。从每个受试者身上选取不同的暴露皮肤部位进行微生物样品采集,采样部位为:潮湿(肘前窝)、干燥(手掌和前臂掌侧)和皮脂腺(关节后皱褶),代表不同的生理特征[27]。使用“拭子-刮擦-拭子”方法收集皮肤微生物,用缓冲液(0.15 mol/L氯化钠 和 0.1% Tween-20)润湿后的拭子对限定的浅表皮肤区域(2 cm × 2 cm)擦拭40 s,用无菌一次性玻璃片轻轻刮擦,再次用相同的棉签擦拭收集,并从玻璃片上收集残留的刮屑[26,28-29]。将来自暴露皮肤部位的样品混合到一个皮肤样品中,以代表暴露的人体皮肤。所有样本保存在4℃并运送到实验室,并在24 h内完成DNA提取。
1.2 方法
1.2.1 DNA提取 水样经真空过滤装置将样品富集在0.22 μm孔径的滤膜上,滤膜用无菌剪刀剪碎后,使用商业试剂盒(FastDNA SPIN Kit for soil, MP Biomedicals, Santa Ana, CA, USA)提取基因组DNA。所有粪便样品在-80℃下冷冻干燥后研碎,使用商业试剂盒进行DNA提取。皮肤样品由蛋白酶K在55℃下孵育预处理6 h,加入978 μL磷酸钠缓冲液和122 μL Tween-20缓冲液,样品涡旋混合3 min,将样品富集在0.22 μm孔径的滤膜上,滤膜用无菌剪刀剪碎后使用商业试剂盒提取DNA。通过分光光度计分析(NanoDrop ND-2000c,Thermo,USA)确定纯化DNA的质量和浓度,并在-80℃下储存直至需要。
1.2.2 高通量荧光定量PCR检测ARGs 使用296对引物在TaKaRa SmartChip高通量荧光定量PCR系统上检测ARGs和MGEs[16]。其中285对引物检测主要抗生素类别的ARGs,包括对氨基糖苷类、β-内酰胺类、氟喹诺酮类/喹诺酮类/氟苯尼考/氯霉素(FCA)、大环内酯类/林可酰胺/链霉素 B(MLSB)、磺胺类、四环素、万古霉素和其他类抗性基因,9对引物检测转座酶基因,1对引物检测对I类整合子基因和1对引物检测16S rRNA基因。所有高通量荧光定量PCR均一式3份进行检测,过程为:(1)在95℃下初始变性10 min;(2)在95℃下30 s变性;(3)在60℃下退火30 s;进行40个循环。最后软件自动生成熔解曲线分析。数据质量控制条件为:(1)效率在(90%-110%)范围内;(2)扩增子无多重熔解曲线;(3)3次重复均在检测限内(阈值循环(Ct)为31)。基因相对丰度计算如下:
1.2.3 细菌16S rRNA 基因测序 使用通用引物515F/907R(GTGCCAGCMGCCGCGG/CCGTCAATTCMTTTRAGTTT)在Ion Torrent平台上扩增细菌16S rRNA基因的V4-V5区域[30]。使用独特的七核苷酸条形码标记每个样品的引物组,以在单次焦磷酸测序运行中识别混合物中的单个样[31-32]。每个样本的所有序列都经接头序列过滤、筛选去除低质量读数和不明确的序列。使用QIIME生成和分析高质量序列,使用UCLUST以97%的相似性水平将序列聚类为操作分类单元(OTU)。
1.2.4 数据分析 基本统计数据分析在Microsoft Excel上进行。使用SPSS V22.0(IBM, USA)分析Spearman 相关系数,显著性水平为P< 0.05。使用ANOVA比较不同样品之间的ARGs丰度差异,显著性水平为P< 0.05。使用ANOSIM比较细菌群落间的差异性。使用LEfSe来分析识别在人类粪便、皮肤和水中差异性细菌[33],基于≥4.0的线性判别分析(LDA)效应大小阈值。基于Bray-Curtis距离的Mantel test和Procrustes分析细菌群落和ARGs间的相关性。使用Gephi平台和Fruchterman Reingold算法对网络进行可视化,只有统计上稳健的相关性才用于形成最终的网络图[13,34-35]。
2 结果
2.1 ARGs分布和共享特征
ARGs检出数量从高到低依次为,粪便>污水>河水>皮肤>自来水,分别检出了223、176、155、140和128种ARGs(图1)。ARGs相对丰度平均值从高到低依次为,粪便>污水>皮肤>自来水>河水,相对丰度为7.88E-03、5.86E-03、1.93E-03、9.19E-04和6.46E-04 copies/16S rRNA 基因。ARGs相对丰度中位值从高到低依次为,皮肤>污水>自来水>粪便>河水,相对丰度分别为1.14E-03、2.66E-04、2.14E-04、5.09E-05和3.39E-05 copies/16S rRNA基因。人体和水环境样品之间共同检出了70个基因,这些被称为共享ARGs。热图显示了共享ARGs的相对丰度(图2)。环境中共享ARGs未被水处理系统完全去除,且与人体肠道和皮肤中的ARGs存在共享,说明了共享ARGs可以在不同介质中传播扩散,可能间接反映了区域内抗生素耐药风险水平。在共享ARGs中,β-内酰胺、MLSB和四环素类的ARGs在人类粪便中占主导地位,而氨基糖苷类和其他类的ARGs在自来水、皮肤、污水和河流样本中占主导地位(图3)。
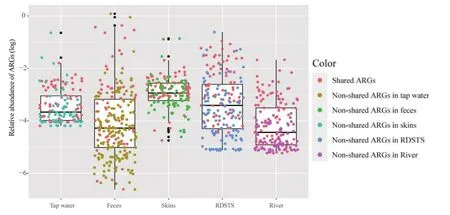
图1 人体和水环境样品中抗生素抗性基因相对丰度Fig.1 Relative abundances of ARGs in humans and water samples
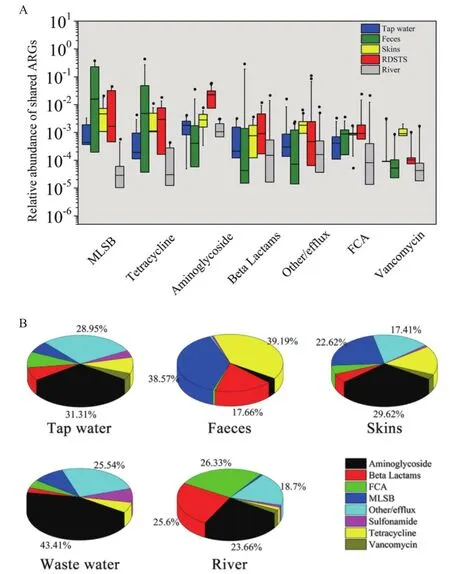
图3 不同环境中共享 ARGs 的相对丰度(A)和比例(B)Fig.3 Relative abundances(A)and proportions(B)of shared ARGs in different environments
2.2 细菌群落分布和共享
经过组装和质量过滤后,从所有样本中总共获得了1 078 819个高质量序列。通过以97% 的不同程度对OTU进行聚类,总共获得了7 277个OTU。粪便、皮肤和水样中细菌群落的组成存在显著不同(ANOSIM test,R= 0.757 9,P< 0.001)。LEfSe结果显示人类粪便、皮肤和水环境的微生物特征之间存在统计学上的显著差异(P< 0.001)(图4-A)。拟杆菌门和厚壁菌门是人类粪便中的优势门,占总细菌16S rRNA基因序列的89.5%。放线菌和变形菌是人体皮肤样本中的主要门(69.3%),而拟杆菌和变形菌在水环境中占优势(70.2%-86.1%)(图4-B)。在科水平上,来自4个门(放线菌门、拟杆菌门、厚壁菌门和变形菌门)的20个分类群在科类别之间存在差异(α < 0.05,LDA ≥ 4.0;图4-C)。人类粪便中的微生物群落在4个不同科中的比例显著较高,包括拟杆菌科、瘤胃球菌科、毛螺菌科和韦荣菌科(图4-C)。人体和水环境样品之间在属水平上共同检出了58个细菌,这些细菌被称为共享细菌。热图显示了共享细菌的相对丰度(图5)。共享细菌的总丰度大小依次为粪便>皮肤>河流>自来水>污水,分别占总细菌的78.0%、64.0%、33.8%、33.3%和29.0%。共享细菌的平均相对丰度大小依次为粪便>皮肤>河流>自来水>污水,分别占总细菌的1.3%、1.1%、0.6%、0.6%和0.5%。共享细菌的中位值相对丰度大小依次为自来水>皮肤>河流>污水>粪便,分别占总细菌的0.16%、0.07%、0.06%、0.05%和0.01%。在共享细菌门水平上中,拟杆菌门在人类粪便中占主导地位,放线菌门和变形菌门在人体皮肤中占主导地位,厚壁菌门和变形菌门在自来水中占主导地位,变形菌门和拟杆菌门在污水和河流样品中占主导。在共享细菌属水平上,人体粪便中丰度最高的是拟杆菌门的Bacteroides,人体皮肤中丰度最高的是放线菌门的Propionibacterium,自来水中丰度最高的是厚壁菌门的Ruminococcus,污水中丰度最高的是变形菌门的Zoogloea,河流中丰度最高的是拟杆菌门的Flavobacterium。共享的细菌可以在不同环境中扩散和定植,可能具有较大的生态位优势。Procrustes 分析中共享ARGs和共享细菌群落间存在显著相关性(Bray-Curtis,R= 0.894 6,P< 0.001,number of permutations = 9 999).Mantel test中共享ARGs和共享细菌群落间也存在显著相关性(Bray-Curtis,R= 0.353 3,P= 0.001 8, number of permutations= 9 999)。
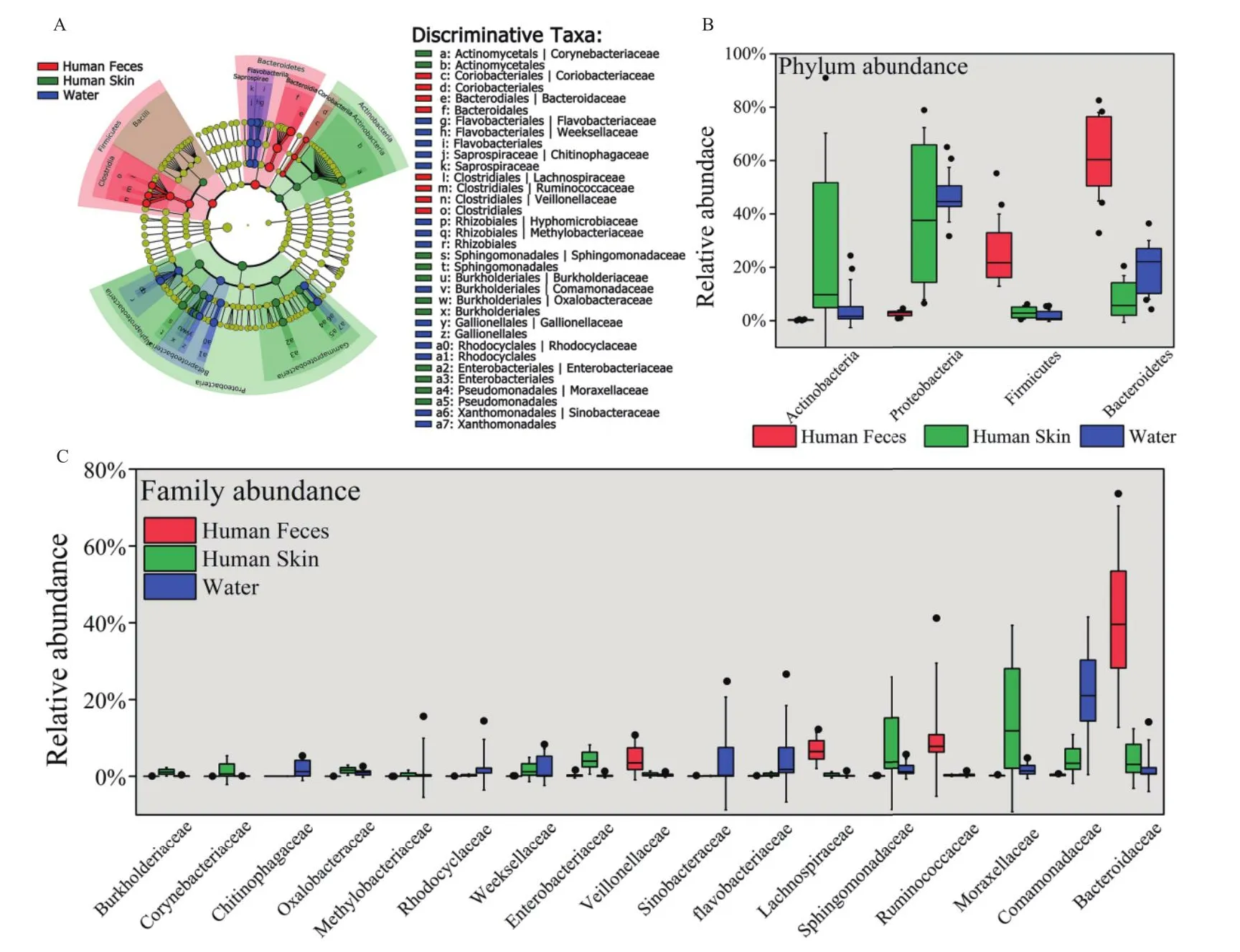
图4 基于LEfSe区分人类粪便、皮肤和水环境中差异特征微生物分类群Fig.4 Taxa discriminates among human feces, skin and water microbiota as determined by LEfSe
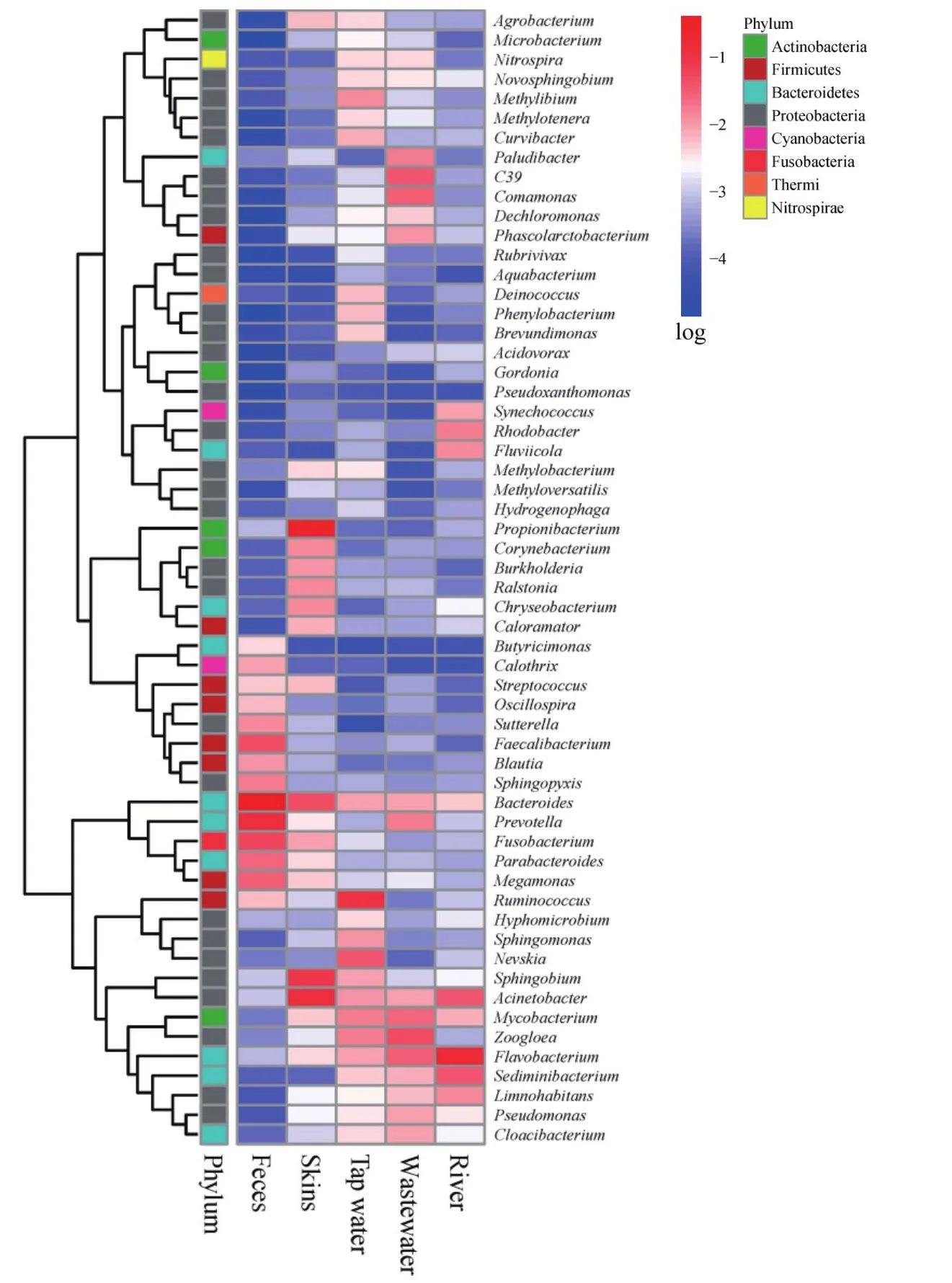
图5 共享细菌丰度热图Fig.5 Heatmap of shared bacteria abundances
2.3 共享ARGs、MGEs和细菌的共出现网络分析
基于强(Spearman 相关系数R> 0.7)和显著(P< 0.01)相关性,通过网络分析分别探索了所有ARGs、MGEs和细菌的共现模式,称为整体网络(图6-A),和70个共享ARGs、7个共享MGEs和59个细菌的共现模式,称为共享网络(图6-B)。整体网络的可视化包括312个节点(每个节点代表一个ARGs、MGEs或细菌子类型)和2 054条边,平均度为13.167,平均加权度为22.336,网络直径为8,图密度为0.042,模块化系数为0.453,平均聚类系数为0.284,平均路径长度为3.045。共享网络的可视化包括132个节点(每个节点代表一个共享的ARGs、MGEs或细菌子类型)和1 053条边,平均度为16.076,平均加权度为26.543,网络直径为6,密度为0.124,模块化系数为0.377,平均聚类系数为0.554,平均路径长度为2.64。共享网络的平均度、平均加权度、图密度高于整体网络,说明了共享ARGs、MGEs和细菌间的交流传递比较高效。整体网络的平均聚类系数低于共享网络,而平均路径长度大于共享网络,说明了整体网络更具有小世界性。共享网络可以清楚地分为4个模块,其中两个最大的模块(模块 I 和 II)占据了132 个节点中的73个。同一模块中的共享基因和细菌可能比其他模块中的更频繁地相互作用和交流,并在人类和水环境中表现出相似的命运。网络分析中,每个节点的大小与连接数成正比,每个模块中连接最密集的节点被定义为“hub”。共享网络中,氨基糖苷类抗性基因aacA4-01是模块I的hub,外排泵介导的多重抗性基因mtrC-02和tolC-02是模块II和模块IV的hub,I类整合酶基因intI是模块 III 的hub(图3)。这些hub类基因在模块中与其他基因和细菌间的连接更加紧密,可以作为典型ARGs和MGEs进行主要关注。
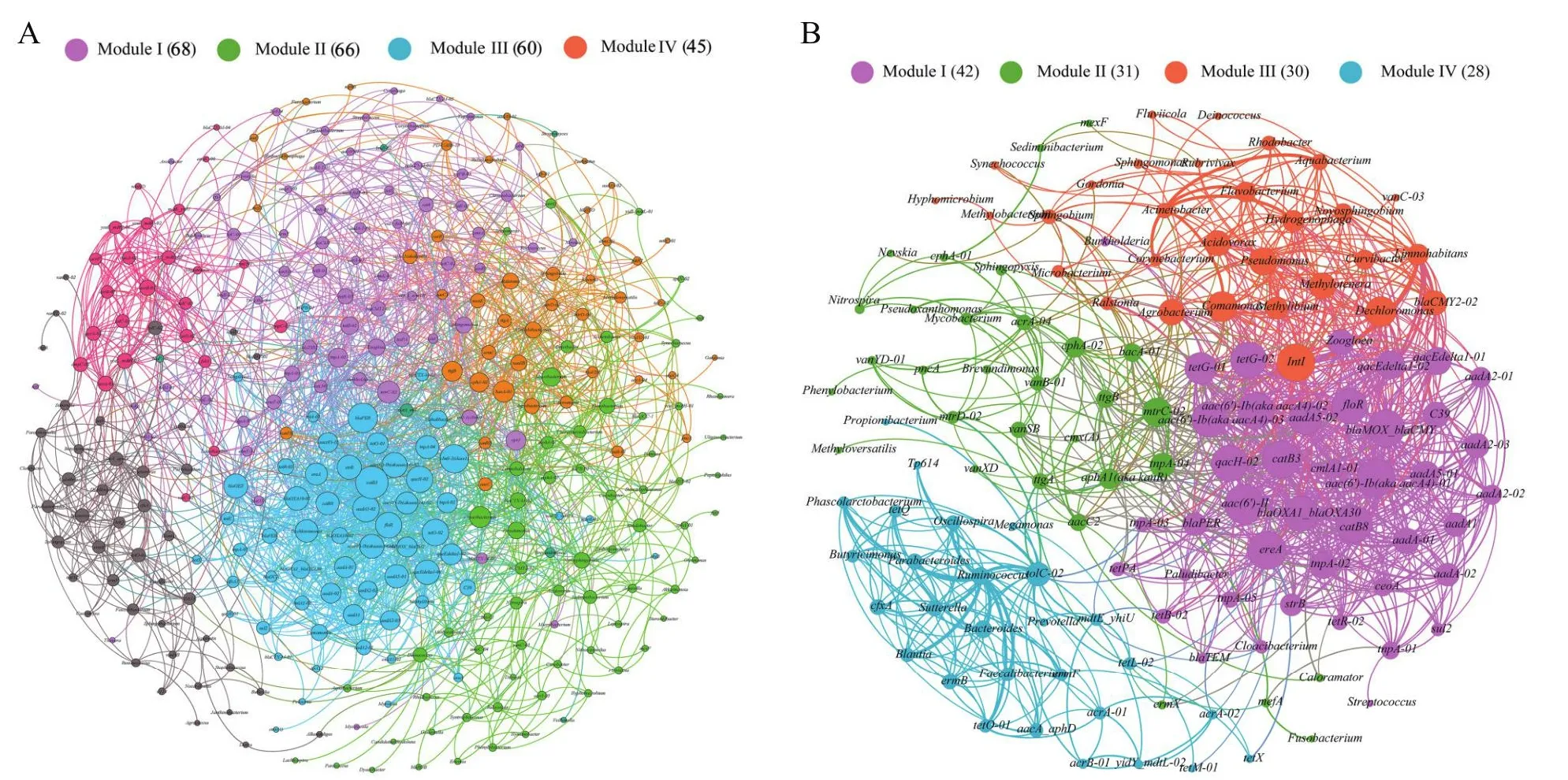
图6 所有检测到的 ARGs、MGEs和细菌的网络分析(A)和共享 ARGs、MGEs和细菌的网络分析(B)Fig.6 Network analysis of all detected ARGs, MGEs and bacteria(A)and shared ARGs, MGEs and bacteria(B)
3 讨论
污水中ARGs在数量和丰度上都较高,之前的研究也表明常规污水处理工艺不能完全去除ARGs且可能导致污水中ARGs的丰度上升,说明了污水可能是ARGs在城郊环境中增殖的主要场所和传播的主要渠道[5,36]。以“人体-污水-地表水-饮用水-人体”这一路径思考,人体肠道和皮肤上的ARGs经生活污水进入污水处理系统。污水系统内接收了大量的生活污水,且未能被污水处理系统去除的ARGs将被释放到地表水中,已有研究也发现了污水处理系统出水排放至河流中将导致河流中ARGs变化[5,36]。河流从人类活动中接收到多种污染物,而且地表水中共享ARGs可能会传播至饮用水水源,例如下游地表水与鱼类间存在共享ARGs[22],而鱼类可以在河流和水库等在不同水域栖息,可能会造成ARGs传播至饮用水水源。已有研究也报道了饮用水处理系统也不能完全去除ARGs[2-4],而且自来水中存在ARGs可能受余氯[2]和输送管道中的生物膜影响[17],自来水对人体的暴露量比污水和地表水更高,其中共享ARGs可能会更容易传播至人体带来潜在的健康风险[37]。β-内酰胺抗性基因在人类和水环境中共享存在,β-内酰胺是全球消耗量最大的三大抗生素之一[38],这类ARGs的广泛传播可能会对治疗细菌感染造成较大的影响。人类粪便中β-内酰胺类、MLSB和四环素类ARGs的富集可能与消耗大量抗生素和杀菌剂有关。细菌携带的这些ARGs可能在多种环境中对细菌的生存具有重要作用,人体内的细菌可能会受到较高的抗生素压力,而这些携带ARGs的细菌比不耐药的细菌具有竞争优势,更易在人体中的生存和定植。此外,据报道MLSB、β-内酰胺类和四环素类是养殖行业主要使用的抗生素[39],人类粪便中这些ARGs的富集也可能是由于自牲畜消费过程中的抗生素、ARGs或者耐药细菌传播至人体。污水中具有丰富的共享ARG可能是因为抗生素、天然抗生素和复杂污染物的残留可能会影响细菌群落和ARGs,细菌携带的ARGs可能会赋予细菌应对不同环境压力的能力,而且污水也可能会促进ARGs在细菌间的水平转移[40]。人类皮肤和水样中丰度高的3个科是Comamonadaceae、Moraxellaceae和Sphingomonadaceae,与粪便有显著差异。这些显著差异分类群可以作为Biomarkers[33,41-42]代表人类和水环境中的不同微生物群落。细菌群落的变化可能会影响ARGs的变化,已有研究也报道了水环境中细菌群落改变可能会对ARGs结构具有一定影响,影响程度在19.65%-68.44%[16,35,43]。整体网络的模块化系数值大于0.4,表明整体网络具有显著的模块化结构,而共享网络不具备模块化结构间接说明了共享网络节点间的联系可能更加紧密[44]。网络分析也可以揭示潜在的共享ARGs宿主,例如,模块III中的Pseudomonas与本模块内的intI-1和blaCMY2-02,与模块I中的aacA4-03、qacH-02、catB3、aadA5-02、blaMOX_blaCMY,与模块II中的mtrC-02存在显著相关性(R= 0.75-0.91,P< 0.01),说明了Pseudomonas可能是这些基因的潜在宿主。Pseudomonas是常见的条件致病菌并且广泛分布在环境和人体微生物群落中,而blaCMY2-02编码的β-内酰胺酶可以使细菌对广谱头孢菌素等β-内酰胺抗生素产生抗性,更为重要的是水平基因转移元件intI与Pseudomonas和blaCMY2-02存在显著相关性并在同一个模块中出现,说明了intI可能会促进这类ARGs在细菌间的转移和扩散。世界卫生组织也报道了对碳青霉烯等β-内酰胺抗生素具有抗性的Pseudomonas aeruginosa是威胁人类健康的最关键的临床抗生素抗性细菌(Priority 1:Critical)[45]。之前的研究也使用网络分析鉴定了地表水和污水处理系统中ARGs的潜在微生物宿主[26,46],确定人体和水环境之间共享ARGs、MGEs和细菌的共出现网络可能有助于识别和控制关键的ARGs亚型在城郊区域传播。
4 结论
抗生素抗性基因越来越需要依据“One Health(大健康)”框架,在人与自然的不同介质中进行综合地表征和定量。本文通过高通量qPCR技术揭示了环境与人体中抗生素抗性基因分布特征和潜在宿主。在人体和水环境中的共享ARGs表现出多样的抗生素抗性类别和较高的基因丰度,而且ARGs的分布与微生物群落存在显著的相关性。ARGs和细菌的网络分析揭示了共享ARGs、MGEs与细菌的共出现和传播,揭示了共享ARGs的潜在细菌宿主。结果为鉴定和识别环境中潜在高风险的ARGs提供了一定的科学依据。
猜你喜欢
数学小灵通(1-2年级)(2023年1期)2023-02-10 06:00:48
娃娃乐园·综合智能(2022年7期)2022-07-16 03:54:36
疯狂英语·新悦读(2021年10期)2021-11-23 03:04:01
中老年保健(2021年6期)2021-08-24 06:53:34
学生天地(2020年14期)2020-08-25 09:20:56
小读者(2019年20期)2020-01-04 02:13:58
南方周末(2019-12-05)2019-12-05 11:17:43
家庭医学(下半月)(2019年9期)2019-10-12 08:03:52
国外医药(抗生素分册)(2016年1期)2016-07-10 12:02:35
发明与创新(2016年33期)2016-04-16 16:32:25
