
金属相二硫化钼催化剂的液相制备及其析氢性能研究
2023-08-10 02:27任浩文陈世宏罗春晖闫康平
电子元件与材料 2023年6期
任浩文,陈世宏,罗春晖,赵 强,闫康平
(四川大学 化学工程学院,四川 成都 610000)
二硫化钼(MoS2)因其特殊的二维结构和化学、物理特性而被广泛研究。尤其是在电解水析氢反应方面,MoS2被证明具有与铂(Pt)相近的理论氢原子吸附自由能[1],因此被认为是一种能够替代贵金属的优质催化材料。但从客观角度出发,MoS2作为析氢催化剂,其催化活性与贵金属还存在一定差距,因此研究如何提升其催化活性具有相当重要的意义。在地球上,MoS2几乎都以热力学相对稳定的半导体相(2H-MoS2)存在。然而,目前的研究表明,在2H-MoS2纳米片层中,仅边缘配位不饱和的原子具有理想的催化活性,而处于基面的原子几乎不具备化学活性。同时,对于二维片层结构来讲,边缘处暴露的活性位点数量非常有限,因此会极大制约材料的整体催化性能[2,3]。此外,由于其本身是半导体,电子传输能力较差,也会限制催化过程的进行[4-5]。与2H-MoS2相比,1T-MoS2不仅边缘位点具有理想的催化活性,其基面也具有较好的催化活性。因此,1T-MoS2的催化活性位点数量可以明显增加。与此同时,1T-MoS2还具有像金属一样的导电能力,其导电率比2H-MoS2高出五个数量级[6-7]。已经有多项研究表明,在合成二硫化钼的过程中,通过各种手段增加1T-MoS2的比例来增加催化剂的整体催化活性。目前已经报道的手段主要是通过碱金属离子插入2H-MoS2层间,通过化学剥离的手段,制备单层1TMoS2[8-9]。然而,这个手段存在反应周期长、产率较低、合成方法复杂和产物不稳定容易再转化为2HMoS2等问题,因此不适合大规模工业应用[10-11]。
针对上述提到的问题,本文提出了一种采用酸性溶剂体系为基础的水热法合成富含1T-MoS2的方法。通过向水热体系中加入无机酸和有机酸,改变硫源和钼源之间的反应合成路径,发现使用有机酸可以提高1T-MoS2的产率。据此推断,在酸性溶剂中硫脲具有更强的还原性,能够将钼酸根中的Mo 还原从而产生四价Mo,并与氧原子形成八面体配体。随后,经过硫化反应转化为具有八面配位结构的1T-MoS2。本文分别使用盐酸、甲酸和乙酸作为酸性溶剂,发现不同酸能够在一定程度上改变1T-MoS2的含量,并证明产物的催化活性随着1T 相的含量升高而升高。最终发现用乙酸作为酸性溶剂得到的产物具有高达75%的1TMoS2占比。特别地,与甲酸作为酸溶剂合成的材料相比,乙酸的碳链相对较短,可以均匀地覆盖每个二硫化钼分子,另一端的疏水官能团可以有效地防止二硫化钼的聚集,避免了1T 相向2H 相的再转化,从而避免了材料降低析氢活性。MoS2-A 在0.5 mol/L 的硫酸中表现出了优异的催化活性。在10 mA/cm2的电流密度下,该催化剂对应的过电位为324 mV,Tafel 斜率为95 mV/dec。通过对三组材料进行物性表征和电化学分析比较,MoS2-A 催化剂优异的催化性能源于1T-MoS2含量高,从而能够提供丰富的催化活性位点以及优异的电子传输能力。
1 实验
1.1 材料合成
本文所用到的催化剂均采用简单水热法合成,示意图如图1 所示。首先称取钼酸铵44.27 mg,硫脲121.73 mg,再将两种药品溶解于30 mL 去离子水中,放入搅拌子在磁力搅拌器上搅拌30 min 至充分溶解后,分别用盐酸、甲酸、乙酸将混合液pH 值调至2后,将上述混合液置于容积为50 mL 的水热釜内衬中,在180 ℃条件下水热反应10 h 得到黑色混合溶液。离心收集反应得到的纳米材料后,分别用乙醇和去离子水清洗三次至中性,冷冻干燥12 h 后将所得材料在900 ℃、Ar 条件下保温2 h,升温速率为5 ℃/min,研磨均匀后将最终所得催化剂命名,按照上述使用酸的顺序分别命名为MoS2-H、MoS2-F、MoS2-A。

图1 MoS2合成示意图Fig.1 Schematic illustration of the main synthesis of MoS2
1.2 电化学测试
本文的电化学测试所得数据均通过CHI760E 电化学工作站获得。测试采用以石墨棒为对电极(CE),填充饱和硫酸钾的汞/硫酸亚汞(Hg/Hg2SO4)为参比电极(RE),直径为5 mm 的玻碳电极为工作电极(WE)的标准三电极体系。其中汞/硫酸亚汞参比电极的电势采用标准可逆氢电极在0.5 mol/L 的硫酸电解液中校准得到(E(RHE)=E(Hg/Hg2SO4)+0.656 V+0.059·pH)。
工作电极的制备: 首先将玻碳电极用Al2O3粉末(50 nm)抛光打磨至测试所需要的表面洁净度,然后配置催化剂ink: 将5 mg 所制备的催化剂加入到200 μL 乙醇、250 μL 和50 μL Nafion(质量分数5%)的混合溶剂中,超声30 min 至充分分散。此后将所得ink均匀滴涂在玻碳电极上,工作电极的催化剂负载量约为0.383 mg/cm2。
电化学测试条件: 在所有电化学测试开始之前,先向电解液中通入氩气30 min,使之后的电化学测试都是在饱和氩气的状态下进行。电极的活化采用循环伏安扫描法(CV),直至得到稳定的CV 曲线;线性伏安扫描法(LSV)在3 mV/s,1600 r/min 的条件下测试得到;通过双电层电容(Cdl)对材料的活性表面积进行表征,通过在0.1~0.2 V(vs.RHE)电位区间、扫速分别为10,20,30,40 和50 mV/s 条件下拟合得到;电化学阻抗(EIS)则是在频率0.01~10000 Hz 范围下测试拟合得到。
1.3 材料表征
X 射线衍射(XRD)数据通过衍射仪(日本Rigaku Smartlab)在40 kV 和40 mA 条件下,从10°到80°,扫速为5(°)/min 时得到;X 射线光电子能谱(XPS)数据通过能谱仪(美国Thermo Scientific K-Alpha)使用Al靶在12 kV 和6 mA 条件下测试得到;场发射扫描电镜(SEM)则采用双束(FIB-SEM)超高分辨场发射扫描电镜(Helios G4 UC 型)测试得到;拉曼光谱(Raman)通过光谱仪(ThermoFisher DXR2 XI)在激发波长为532 nm,100~500 cm-1条件下测试得到。
2 结果与讨论
2.1 形貌及物性表征分析
利用场发射扫描电镜(SEM)对本实验合成的三种催化剂的形态和微观结构进行表征,如图2 (a,b,c)所示。其中,MoS2-H 表现出尺寸为1~2 μm 的多孔球状结构,由团聚的二硫化钼纳米片组成。采用甲酸调控pH 值的MoS2-F 呈现为尺寸约为1 μm 左右的多孔球状结构,其仍由二硫化钼纳米片堆叠而成。而当采用乙酸调控时,MoS2-A 表现为尺寸为300~600 nm的多孔球状结构,其仍然由二硫化钼纳米片堆叠而成,但堆叠的纳米片数量更多。有机酸可以通过羧酸官能团与二硫化钼连接,促使它们生长成垂直的纳米片,同时阻止纳米片的过度堆叠,暴露出更多的边缘活性位点,其尺寸更小并且拥有更高的比表面积,从而更加有利于析氢反应进行。此外,通用扫描电镜对MoS2-A 进行面扫描元素分析,发现该催化剂中的Mo 和S两种元素分布也非常均匀。
利用XRD 技术进一步表征了所制备的三种材料的结晶度(图3),可以从图中看出,三种材料在14.4°,33.5°,39.5°,58.3°处的峰值可以归结为二硫化钼的(002),(101),(103)和(110)晶面(PDF#37-1492),说明该方法合成的二硫化钼都具备良好的结晶度。值得注意的是,MoS2是最经典的具有二维层状的各相异性材料,其合成过程中优先暴露的是热力学稳定的(002)晶面,而不是活性较高的边缘面[12]。从MoS2-H 到MoS2-A,(002)晶面所对应的峰强度呈现降低趋势,表明其含量相对较低,这可能与MoS2-A 中1T 相占比较高有关,但这还不能完全证明1T 相的产生。为了进一步证明1T 相的产生,采用拉曼光谱对催化剂的结构进行分析评价,如图4 所示。三种材料在388 cm-1和415 cm-1处表现出二硫化钼的标准特征峰,分别对应于MoS2中面内和层间(A1g)这两种分子的振动模式,较高强度的A1g振动模式表明其具有端边结构,因此具有更好的催化活性[13]。值得注意的是MoS2-F 和MoS2-A 在293 cm-1处出现了E1g峰,表明在这两种材料中有1T-MoS2的存在[14-15],同时在153,210,293 和343 cm-1处均出现了J1,J2,J3的明显峰,表明甲酸和乙酸的加入能够在反应过程中促进1TMoS2的产生[16],从而提升催化剂的催化活性,根据峰值强度也能够大概推测乙酸调控pH 值产生的1T 相占比更大,从而具有更高的析氢活性[17]。
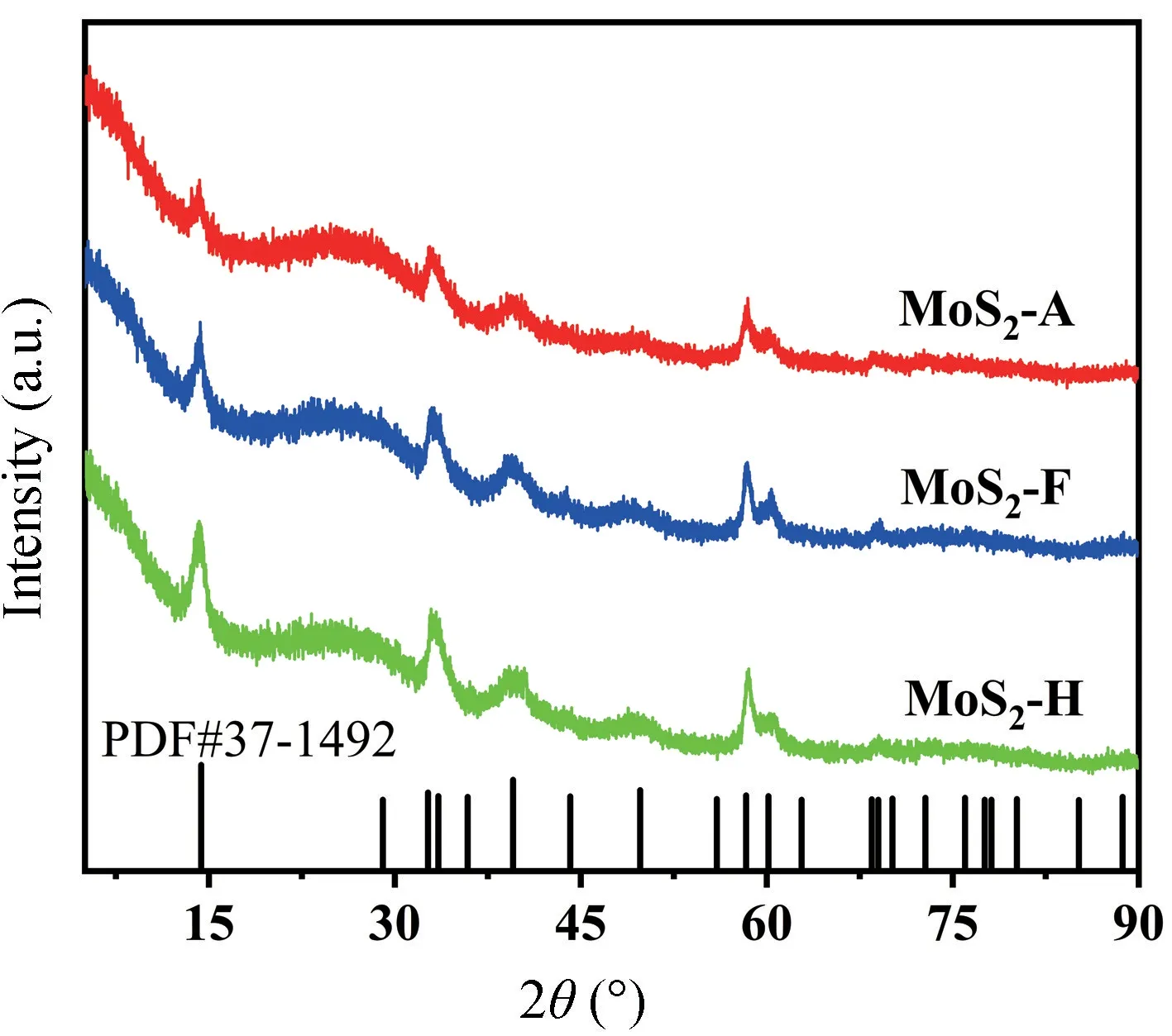
图3 三种不同材料的XRD 图谱Fig.3 XRD patterns of three different materials
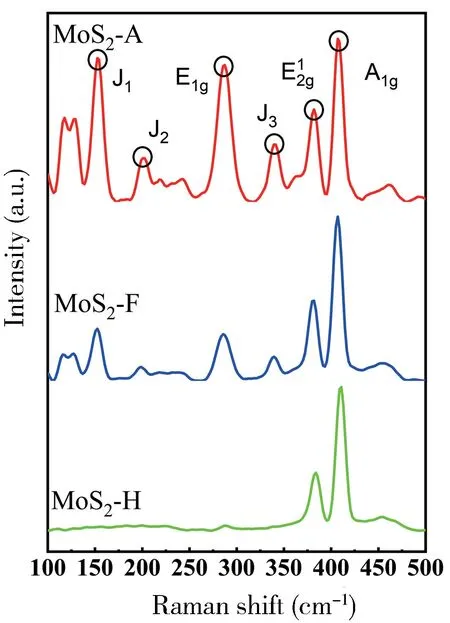
图4 三种不同材料的拉曼分析Fig.4 Raman spectroscopy of three different materials
采用X 射线光电子能谱(XPS)对所制备的三种材料进行化学成分和元素价态分析如图5 所示。由绘制的高分辨谱图发现Mo 3d 轨道有四个峰出现,它们分别出现在228.68,228.98,231.78 和232.18 eV处,其中228.68 eV 和231.78 eV 这两个峰可以识别为1T-MoS2,228.98 eV 和232.18 eV 这两个峰则识别为2H-MoS2[18],同样S 2p 轨道也可以得到相应的结论,除此之外,相对于MoS2-H,MoS2-A 的Mo 3d和S 2p 结合能分别向低结合能方向偏移了0.7 eV 和0.6 eV,表明乙酸组合成的MoS2具备很大的1T-MoS2相占比[19-20]。根据XPS 分峰拟合后的结果,用峰面积计算得到MoS2-A 的1T 相占比高达76%[21]。由此可以证明随着采用甲酸和乙酸替代盐酸调控pH 值,调控了材料的电子结构,得到的二硫化钼具有更多的1T相占比,从而其具有更高的电子传输效率,降低了电解水势垒能,从而提高了析氢性能。
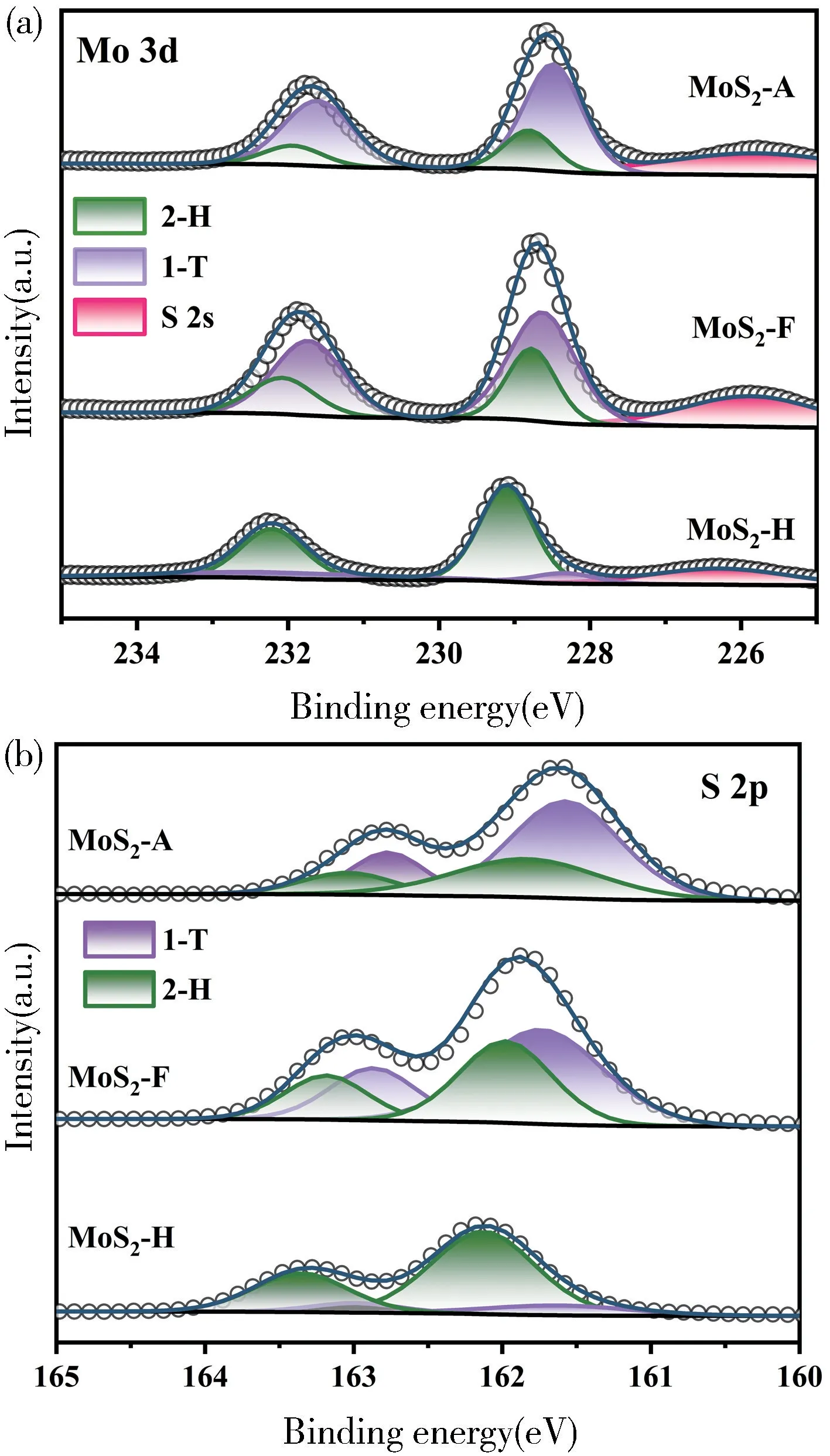
图5 (a) 三种材料的高分辨Mo 3d XPS 谱图;(b) 三种材料的高分辨S 2p XPS 谱图Fig.5 High-resolution spectra of (a) Mo 3d and(b) S 2p of three different materials
2.2 催化剂的HER 性能测试
在对材料的形貌和结构进行表征分析之后,通过线性伏安扫描法和循环伏安法来评价制备的三种材料的电解水析氢性能。首先在0.5 mol/L 的H2SO4中,三电极体系下得到了三种材料的LSV 曲线如图6(a),可以看出MoS2-H,MoS2-F 和MoS2-A 在电流密度为10 mA/cm2的过电位分别为546,531 和324 mV,虽远高于Pt/C 的28 mV,但基于其合成方法简单,成本低廉,依旧具有研究意义。通过拟合LSV 曲线得到了三种材料的Tafel 曲线如图6(b),可以得到MoS2-H,MoS2-F 和MoS2-A 的Tafel 斜率分别为186,161 和95 mV/dec。Tafel 斜率能够体现一个反应的动力学特性,斜率越低表明此材料的催化活性越高,相较于Pt/C 的41 mV/dec,MoS2-A 表现出比较令人满意的动力学活性。
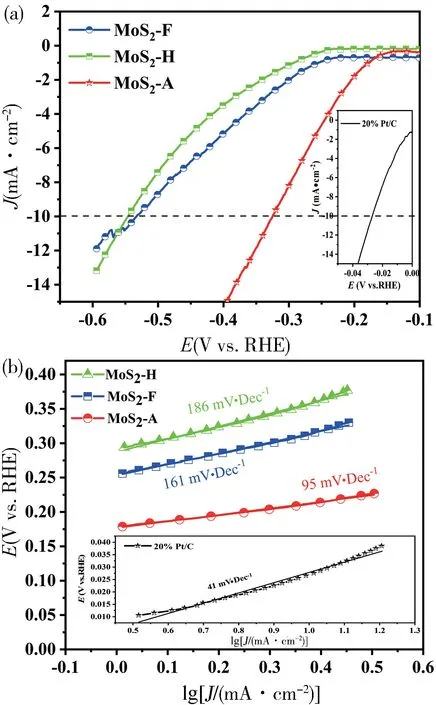
图6 (a)催化剂在LSV 性能曲线;(b)不同催化剂的塔菲尔斜率图Fig.6 (a) The LSV curves of different catalysts;(b) Tafel slope diagram of different catalysts
为了从电化学的角度来解释不同酸合成的材料析氢性能的差异,进行了电化学阻抗分析,绘制的奈奎斯特图如图7 所示。基于等效电路拟合数据,MoS2-A的电荷转移电阻(Rct)约为15.53 Ω,相较于MoS2-F的78 Ω 和MoS2-H 的89 Ω,MoS2-A 在析氢过程中较低的Rct表明其具有更高效的电子转移,这一点可以通过电化学活性表面积(ECSA)来证实,要计算拟合活性表面积就需要通过拟合双电层电容来实现。
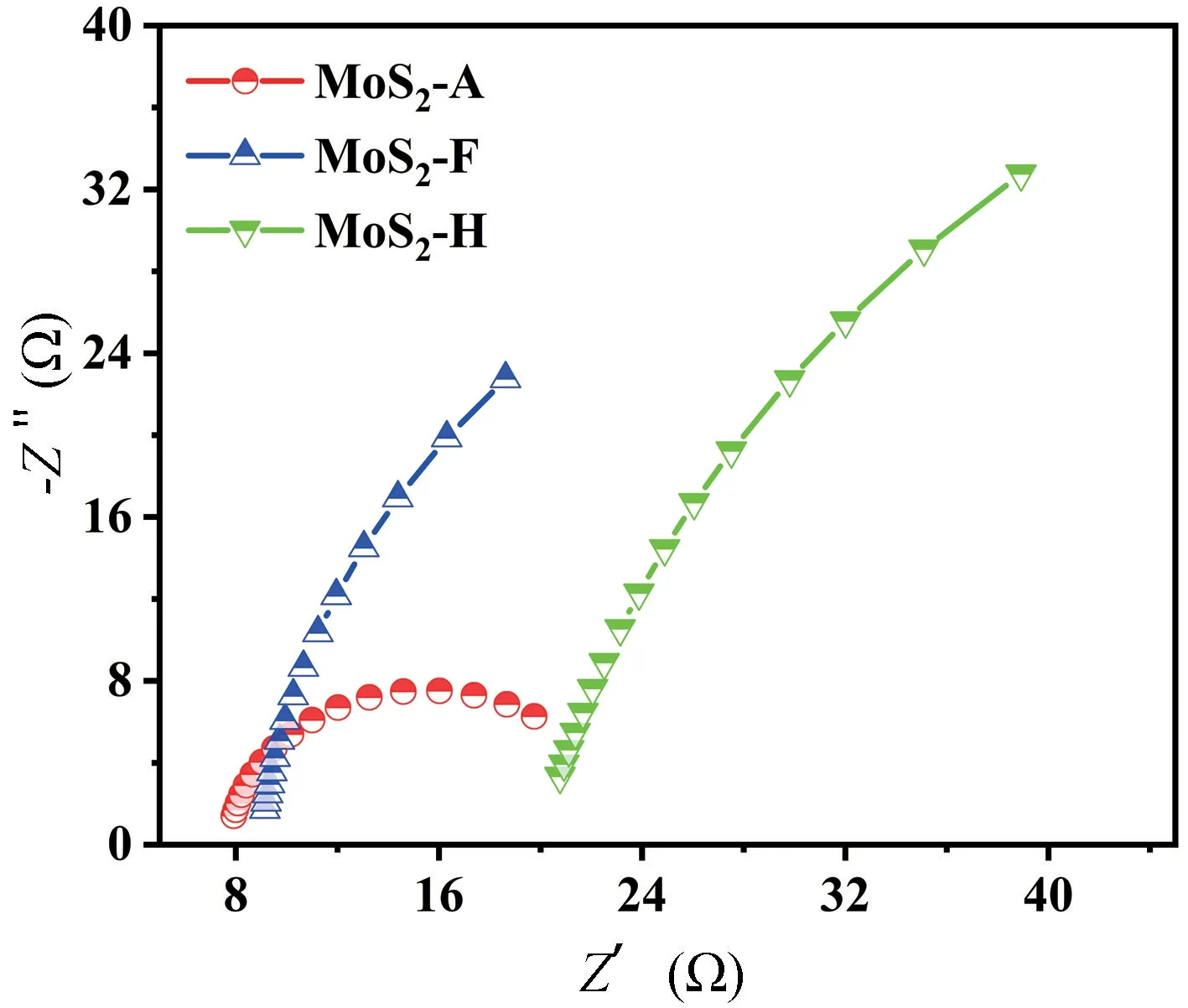
图7 不同催化剂的奈奎斯特图Fig.7 Nyquist plot for different catalysts
通过在非法拉第区间0.1~0.2 V(vs.RHE)范围内采用不同扫速(10~50 mV/s)进行循环伏安测试得到如图8(a)。再对其进行拟合得到不同材料的双电层电容(Cdl)如图8(b),MoS2-A 的双电层电容为0.005 mF·cm-2,高于MoS2-F 的0.003 mF·cm-2,并且远高于MoS2-H 的0.0003 mF·cm-2,由此表明MoS2-A具有最佳的HER 活性位点。这也证明MoS2-A 优异的催化活性源于其具有很大占比的1T-MoS2。
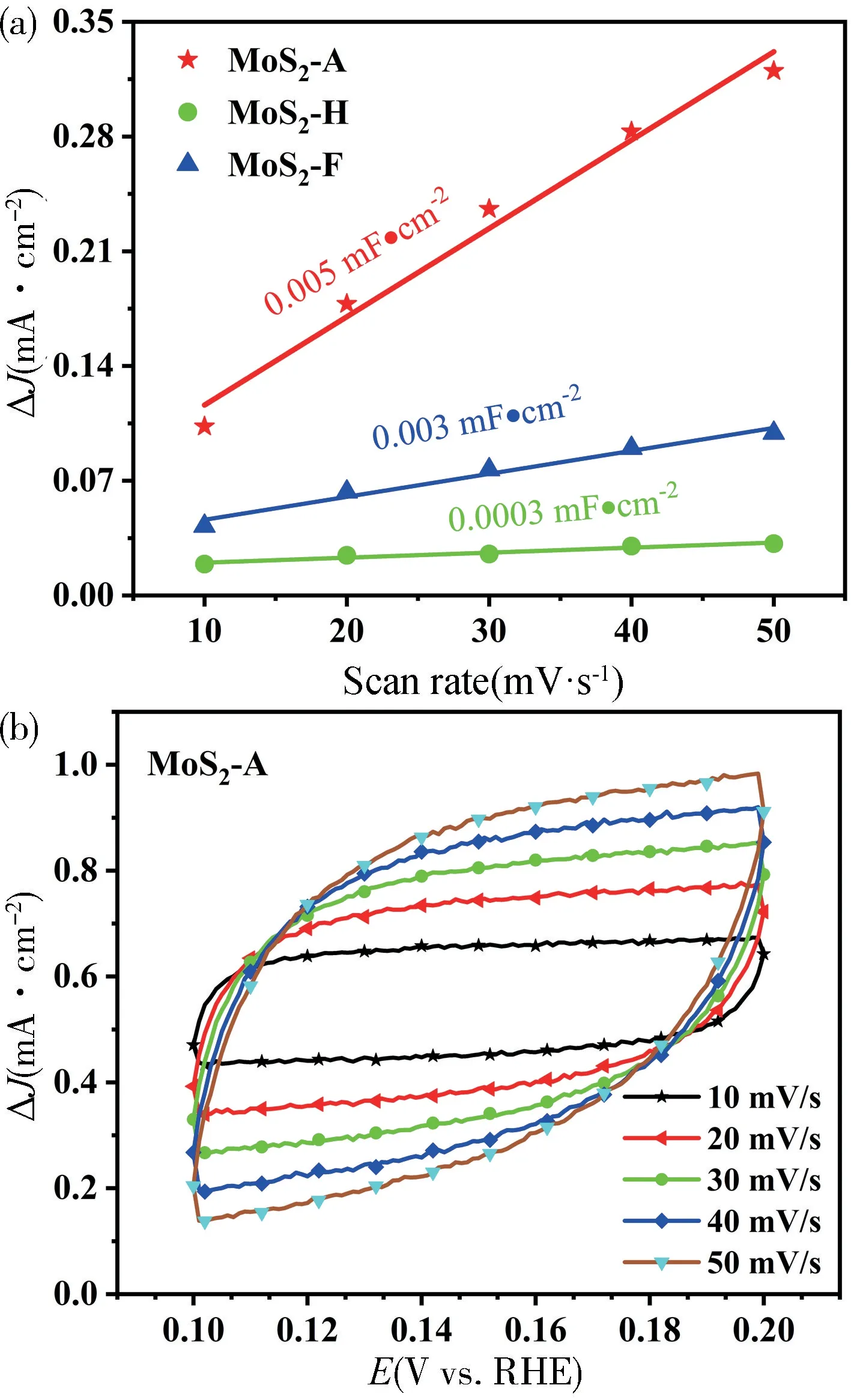
图8 (a)不同材料的双电层容量图;(b)不同扫速下MoS2-A 的CV 图Fig.8 (a)Double layer capacity of different catalysts;(b) CV curves at different scanning speeds
3 结论
在使用水热法合成二硫化钼的过程中,本文发现强酸性水热体系都能够促进1T-MoS2的产生。但是与盐酸和甲酸相比,使用乙酸调控水热反应溶剂能够得到高达76% 1T 相占比的MoS2,并且乙酸的加入可以一定程度上限制纳米片的团聚,同时促进MoS2垂直生长,暴露更多的边缘活性位点,从而获得均匀更小的尺寸和更大的比表面积,更有利于析氢反应的进行。在0.5 mol/L 的硫酸电解液中,MoS2-A 表现出优异的催化活性,其过电位仅为324 mV,在10 mA/cm2的电流密度下,Tafel 斜率为95 mV/dec,电荷转移电阻仅为15.53 Ω,表明1T-MoS2是MoS2-A 优异催化活性的来源,它提供了丰富的催化活性位点以及优秀的电子传输能力。虽然该催化剂的析氢性能与商业Pt/C相比仍有一定差距,但是其合成方法简单,成本低廉的优势使其具有一定的研究价值和应用前景。
猜你喜欢
环境工程技术学报(2022年3期)2022-06-05
中学生数理化·高一版(2022年4期)2022-05-09
陶瓷学报(2021年4期)2021-10-14
表面工程与再制造(2019年1期)2019-05-11
浙江大学学报(工学版)(2016年9期)2016-06-05
当代化工研究(2016年5期)2016-03-20
中国铸造装备与技术(2015年5期)2015-12-10
淮南师范学院学报(2015年3期)2015-03-22
河北科技大学学报(2015年5期)2015-03-11
无机化学学报(2014年4期)2014-02-28
