
Wnt/beta-catenin signaling and its modulators in nonalcoholic fatty liver diseases
2023-08-02 09:00KarthikShreeHariniDevarajEzhilarasan
Karthik Shree Harini, Devaraj Ezhilarasan
Department of Pharmacology, Molecular Medicine and Toxicology Lab, Saveetha Dental College, Saveetha Institute of Medical and Technical Sciences(SIMATS), Chennai, Tamil Nadu 600 077, India
Keywords:Steatosis Fatty liver Oxidative stress Chronic liver diseases Fibrosis Wnt/ β-catenin
ABSTRACT
Introduction
Nonalcoholic fatty liver disease (NAFLD), a spectrum of liver diseases, can be histologically characterized by the accumulation of more than 5% of fat in hepatocytes [1].NAFLD originates with simple steatosis, fat accumulation, progressing through nonalcoholic steatohepatitis (NASH), associated with lobular and portal inflammation, hepatocyte ballooning and necrosis, further progressing to fibrosis, denoting the excess accumulation of extracellular matrix(ECM) proteins with the hepatic stellate cells (HSCs) doing the role and advancing to liver cirrhosis, leading to hepatic insufficiency and portal hypertension and hepatocellular carcinoma (HCC) [ 2 , 3 ].NAFLD is a global concern due to its rapid progress in the last few decades at a rate of 15% in 2005 to 25% in 2010.The overall prevalence of NAFLD contributed to about 25% of the world population by 2018 [4], dominating with 24% in North America, 31% in South America, 32% in the Middle East, 27% in Asia, 24% in the USA, 23%in Europe, and 14% in Africa by 2016 [5].NAFLD is also observed in individuals during adolescence at a rate of 3% to 18% [6].Hence,NAFLD is now becoming one of the major causes of liver-related diseases with significant morbidity and mortality [7].
The host factors having an effect on the origin and furtherance of NAFLD are age, sex, genetics, obesity, metabolic syndrome, type 2 diabetes mellitus and environmental factors.Studies [ 8 , 9 ]have shown that increasing age increases the extent of fibrosis (example: age>50 years).Sex difference is also a factor in determining the NAFLD occurrence rate, and impacting the phenotype.Genetically, the patatin-like phospholipase domain-containing 3 (PNPLA3)single nucleotide polymorphism (SNP), I148M (rs738409 C/G) is found by a genome-wide association study on NAFLD to be correlated with high hepatic fat accumulation and inflammation [10].Also, SAMM50 (sorting and assembly machinery component 50 homolog) and PARVB (parvin beta) SNPs are detected in the development of NASH from simple steatosis [11].NAFLD was documented in 94% of obese persons with a body mass index (BMI)>30 kg/m2by a European study [12].Evidently, NAFLD patients have gained more weight (5.8 ± 6.1 kg than healthy people and 2.7 ± 4.7 kg than those who were treated for NAFLD) [ 13 , 14 ].Some of the identified environmental factors, including high-fat and highsugar diets, obstructive sleep apnea [15]and gut microbial composition [16]increase the risk of NAFLD.
NAFLD is a multifactorial disease including type 2 diabetes mellitus, metabolic syndrome like cardiovascular disease, increased triglycerides (TGs), waist circumference and BMI, hyperglycemia,hypertension, dyslipidemia [17]with the underlying characteristic of obesity and insulin resistance, affecting the extra-hepatic organs [ 1 , 7 ].Metabolic syndrome is one of the main risk factors pertaining to NAFLD, which is bidirectionally associated with each other, triggering either, especially in the case of diabetes and hypertension [18].Metabolic syndrome is also associated with mortality in patients with NAFLD and contributes to the condition leading to cardiovascular abnormalities [19].Metabolic syndrome and type 2 diabetes mellitus, in particular, have an effect on the development of NAFLD [20]rather than NAFLD progression [ 8 , 21 ].It has also been observed that around 70% of patients with type 2 diabetes mellitus potentially develop NAFLD [12].Individuals with diabetes and NAFLD are more likely to develop the advanced stages of NAFLD, NASH, and fibrosis than non-diabetic individuals.Insulin resistance also plays a major role in NAFLD.Insulin resistance promotes the lipase enzyme responsible for the conversion of TG into glycerol and fatty acids and brings them into circulation.This disintegration of lipid homeostasis links diabetes and NAFLD together [22].Insulin resistance has also shown to exacerbate NAFLD advancement [18]and is intricately related to the hepaticdenovolipogenesis that is transcriptionally regulated by the factor SREBP1c (sterol regulatory element-binding protein 1c).Consumption of a high calorie diet upregulates insulin signaling mediated SREBP1c activation that stimulates gene expression responsible for coordinated hepaticdenovosynthesis of TGs.Thus, conditions like insulin resistance and hyperinsulinemia predispose to NAFLD [23].The aim of this review is to summarize the influence of Wnt/βcatenin signaling in NAFLD and discuss on Wnt/β-catenin signaling modulators for the treatment of NAFLD.
Pathogenesis of NAFLD
NAFLD is a worldwide disease with a doubled rate of incidence in males than in females [24]and a low prevalence of fibrosis in children, denoting the less progressive phenotype [25].Pathogenesis of NAFLD and NASH is classically explained as the “twohit hypothesis”, where the “first hit” represents the TG accumulation in hepatocytes, sensitizing the liver to the “second hit” injury by proinflammatory cytokines like tumor necrosis factor alpha (TNF-α) and interleukin-6 (IL-6) and adipokines, resulting in inflammation, oxidative stress, and mitochondrial dysfunction accelerating to steatohepatitis, and fibrosis [26].These cytokines, in turn, maintain the insulin resistance state, contributing to the high levels of TG, fatty acid, free cholesterol, and lipid metabolites [27].In patients with insulin resistance and obesity, insulin resistance aggravates adipocyte dysfunction, results in fatty acid infiltration into the liver via compromised lipolysis inhibition in adipose tissue and escalates hepaticdenovolipogenesis [28].Free fatty acids(FFA) in the liver are subjected toβ-oxidation with glycerol, forming and accumulating as TGs.The “updated two-hit hypothesis”also signifies the direct effect of FFA on liver injury [29]by elevating oxidative stress and abnormal adipose tissue function, producing and secreting inflammatory cytokines and adipokines [30].The healthy liver functions normally by replacing the dead cells with mature hepatocytes, whereas NAFLD patients experience a“third hit”, characterizing insufficient hepatocyte proliferation [31].The hepatocytes replication is inhibited during NAFLD pathogenesis and oxidative stress.The “third hit” is also characterized by the hepatic progenitor cells (HPCs) activation, which could accelerate the progression of fibrosis or cirrhosis, and even HCC [32].
The “multiple-hit” theory in the study of NAFLD is denoted as the network of metabolic dysfunction in various tissues and organs like the pancreas, gut, liver and importantly, adipose tissue [33].The NAFLD pathogenesis begins with either excess fat synthesis, excess fat delivery, lowered fat export or lowered fat oxidation in the liver, leading to the production of lipotoxic species that induce endoplasmic reticulum stress or production of reactive oxygen species (ROS), oxidative stress, and ultimately leads to hepatic inflammation [ 34 , 35 ].Mitochondrial dysfunction and oxidative stress together play a major role in NASH [36].Mitochondrial abnormalities such as mitochondrial DNA exhaustion, morphological and ultrastructural changes are observed in NAFLD.The morphological and functional alterations in mitochondrialβ-oxidation and respiratory chain occur in NAFLD patients [37].With the increased FFA flux, fat homeostasis is dysregulated with excess production of ROS [38].Activation of the inflammatory pathway by ROS intensifies mitochondrial damage.ROS production is also influenced by higher expression of the hepatic microsomal fatty acid oxidizing enzyme (CYP2E1, cytochrome P450 2E1), which was determined using human and animal NAFLD models [39].
Inflammation and progression of fibrosis
The chronic hepatic inflammation in NAFLD is associated with the activation of two inflammatory pathways, c-Jun N-terminal kinase (JNK)-activator protein-1 and Iκκ-βkinase-nuclear factor(NF)-κB [40].Murine models have demonstrated the presence of elevated NF-κB activity, which regulates the expression of TNF-α,IL-6 and IL-1βand the activation of Kupffer cells [41].Iκκ-β/NFκB can directly be activated via FFA flux, adding importance to the inflammation caused due to metabolic syndromes [29].The higher levels of proinflammatory cytokines in hepatocytes lead to NAFLD and to NASH progression, resulting in histological changes like hepatocyte necrosis and apoptosis, neutrophil chemotaxis, activation of HSCs, and Mallory bodies formation [42].Patients with NASH have elevated TNF-αin serum and hepatocyte; similarly, patients with insulin resistance and NAFLD have high levels of serum IL-6 [43].Thus, NF-κB inflammatory pathways promote fibrogenesis and HCC [44].Inflammasome components from injured hepatocytes directly promote HSC activation, which eventually leads to hepatic fibrosis [45].Furthermore, Kupffer cells also activate the inflammasome by IL-1βupregulation in the injured liver [ 46 , 47 ].In a NASH mice model, Toll-like receptor (TLR) signaling promoted IL-1βexpression by Kupffer cells, leading to steatosis, inflammation,and fibrosis [48].
Chronic liver diseases progress from NASH to fibrosis.The quiescent HSCs are accountable for vitamin A storage in a healthy liver.However, HSCs are activated by factors like toxins, inflammatory response, and NASH, acquiring a myofibroblast-like phenotype, which is also called the activated HSCs [49].The activated HSCs express various cytokines and chemokines includingα-smooth muscle actin (α-SMA), endothelin-1, platelet-derived growth factor (PDGF), transforming growth factor (TGF)-βand epidermal growth factor that favor activated HSCs or myofibroblasts proliferation, consequently promoting fibrogenesis [50].Activated HSCs are responsible for enormous ECM synthesis on the chronically injured liver.TGF-β1, a profibrotic cytokine, promotes the accumulation of fibril forming collagen in the space of Disse by inducing the transition of quiescent HSC into myofibroblasts [51].This leads to the capillarization of sinusoids, which advances with the formation of fibrotic septa, vascular changes leading towards cirrhosis, finally resulting in portal hypertension [52].Kupffer cells(CD11b+, F4/80++, CD68+, CXCR1-), the macrophages in liver, are activated by FFA and various other factors and interact with TLRs,which lead to the secretion of interleukins like IL-1β, IL-18, and proinflammatory cytokines and chemokines.Kupffer cells not onlyplay a role in activating HSCs but also destroy hepatocytes.HSCs express chemokine receptors and ligands like CCR5, CCR7, CXCR3 and CCL2, CCL3, CCL5, CXCL1, CXCL8, CXCL9, and CXCL10 that assist the migration of myofibroblasts and enhance the inflammation at the site of injury [53].Other than the Kupffer cells, in humans, CD14+CD16+monocyte-derived macrophages also play a role in activating HSCs [54].Inflammatory cells, including T cells,natural killer cells, dendritic cells, and endothelial cells are also involved in the activation of HSCs [55].The accumulated ECM due to the above-mentioned reasons is preserved by a mechanism where there is an increased expression of tissue inhibitor of metalloproteinases (TIMP) which inhibits matrix metalloproteinases that degrade the ECM [56].These fibrogenic pathways are NASH-specific [57].The severity of fibrosis, repeated apoptosis, and regeneration of hepatocytes leads to cirrhosis.Cirrhosis is also considered the irreversible stage of fibrosis [58].The liver parenchyma is grossly distorted, leading to parenchymal dysfunction.Development of nodules of regenerating parenchymal cells aggravates the condition [59], which may eventually progress to HCC ( Fig.1 ).
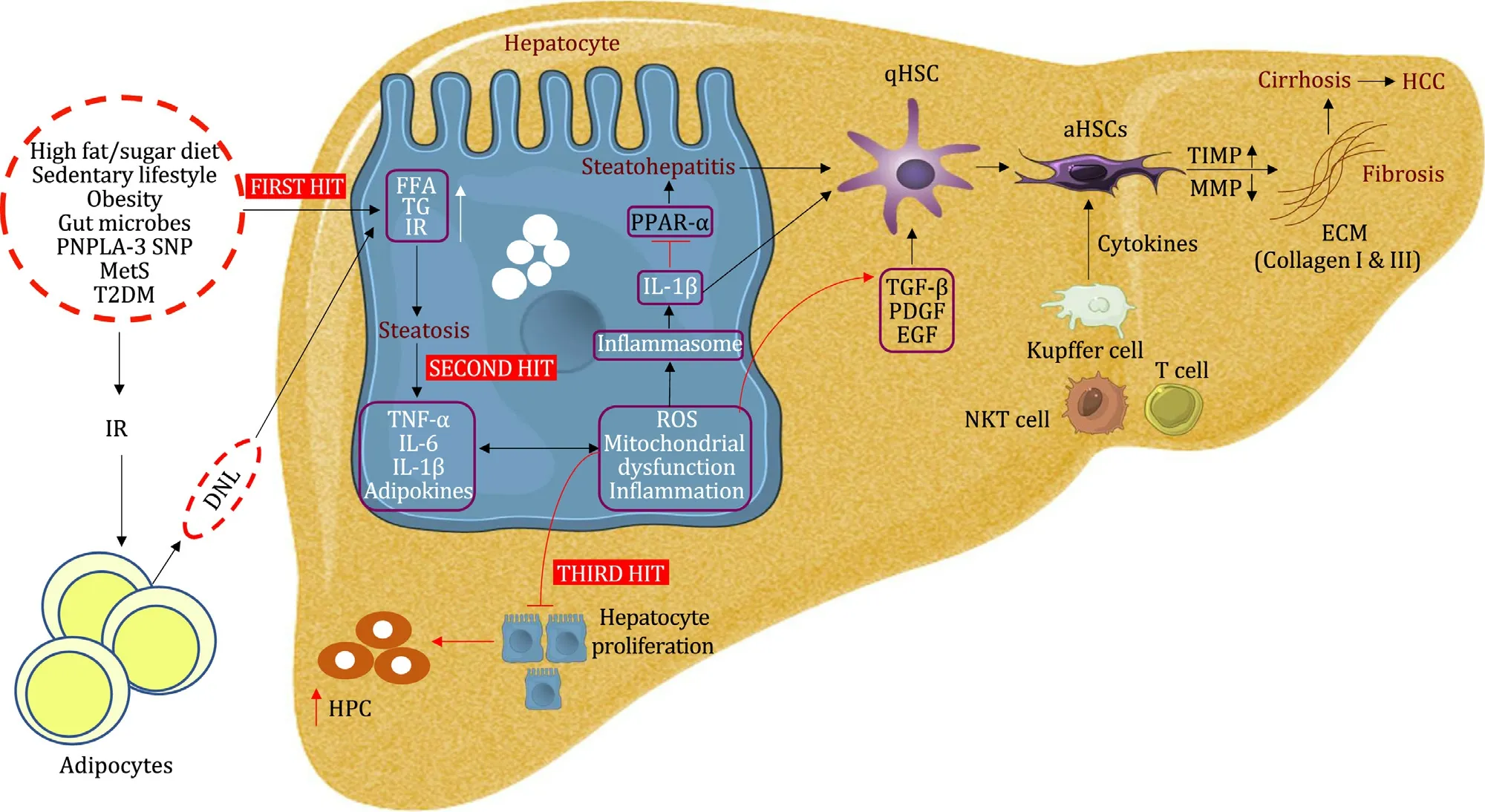
Fig.1.Pathogenesis of NAFLD.The picture shows the progression of NAFLD by first, second and third hits leading to the development of fibrosis, cirrhosis and HCC.The first hit initiates with lipid accumulation, second hit represents the secretion of proinflammatory cytokines and the third hit suppresses hepatocyte proliferation.These responses to liver injury stimulate the HSCs to secrete extracellular matrix protein, resulting in fibrosis that could progress to cirrhosis and HCC.NAFLD: nonalcoholic fatty liver diseases; HCC: hepatocellular carcinoma; HSCs: hepatic stellate cells; PNPLA-3: patatin-like phospholipase domain-containing 3; SNP: single nucleotide polymorphism;MetS: metabolic syndrome; T2DM: type 2 diabetes mellitus; FFA: free fatty acid; TG: triglyceride; IR: insulin resistance; DNL: de novo lipogenesis; TNF- α: tumor necrosis factor alpha; IL-6: interleukin 6; IL-1 β: interleukin 1 β; ROS: reactive oxygen species; HPC: hepatic progenitor cell; PPAR- α: peroxisome proliferator-activated receptor- α;aHSCs: activated HSCs; TGF- β: transforming growth factor beta; PDGF: platelet derived growth factor; EGF: epidermal growth factor; NKT cell: natural killer T cell; TIMP:tissue inhibitors of matrix metalloproteinases; MMP: matrix metalloproteinases; ECM: extracellular matrix.
Wnt/ β-catenin pathway
Wnt signaling is categorized into two different pathways as canonical and non-canonical pathways.The canonical Wnt signaling isβ-catenin dependent.β-catenin is a protein encoded by catenin beta 1 (CTNNB1), functioning as an intracellular signal transducer in the Wnt/β-catenin pathway.β-catenin is responsible for the transcription of genes that are involved in cell proliferation, differentiation, survival, apoptosis, and inflammation mediated by the T-cell factor/lymphoid enhancement factor (TCF/LEF)family of transcription factors [60].This signaling is activated by any of the 19 members (in humans) of the extracellular secreted glycoprotein, Wnt ligand.When Wnt signaling is absent or suppressed by factors like Wnt inhibitors including secreted frizzledrelated proteins (sFRPs), Dickkopf (DKK) and Cerberus,β-catenin bound to a degradation complex is phosphorylated, leading to its proteasomal degradation.The components of the destruction complex, including casein kinase I, glycogen synthase kinase 3β(GSK-3β), adenomatous polyposis coli (APC) gene product, diversin,and axin, phosphorylateβ-catenin at serine-45 (S45), S33, S37,and threonine-41, respectively andβTrCP ubiquitinylates it for its degradation [61].The amount of freeβ-catenin in the cytoplasm is thus maintained very low inhibiting its nuclear translocation and precisely TCF acting as a Wnt-target gene repressor.Contrastingly, the binding of Wnt ligand induces a spatial interaction between the cell surface receptors called frizzled (Fzd) and its coreceptor LRP5/6 (low-density lipoprotein receptor-related protein 5/6), forming a ternary complex [62].The Fzd receptors belong to the class F superfamily of G protein coupled receptors (GPCRs)with a principle organizational structure containing an extracellular N-terminus and an intracellular C-terminus with seven transmembrane helixes connected by three extracellular and three intracellular loops [63].Wnt undergoes glycosylation and acylation to become bioactive, aided by a porcupine (PORCN) (a membranebound O-acyltransferase) to bind to Fzd receptor [64].The ligand binding causes conformational changes in the receptor, resulting in the activation of G protein by replacing GDP with GTP at Gαsubunit, which in turn is responsible for the activation of Wnt downstream signaling.Upon the activation,β-catenin is stabilized in its monomeric form, avoiding its destruction facilitated by the activation of Dishevelled (Dvl) [65].Eventuallyβ-catenin is translocated to the nucleus, triggering the Wnt target genes expression.Thusβ-catenin stimulates the transcriptional ability of TCF4.
Wnt/ β-catenin pathway in healthy liver
Wnt/β-catenin signaling is one of the many pathways required for the normal liver prenatal development, postnatal growth,regeneration, hepatic metabolic zonation, ammonia and drug detoxification, hepatobiliary development, with overall maintenance of adult liver metabolism and homeostasis.In the earlier stage of liver development,β-catenin is found in the hepatocyte membrane, cytoplasm, and nucleus, and is responsible for the proliferation of immature hepatoblasts [66].In the later stages,truncatedβ-catenin is primarily found in the hepatocyte membrane and in a few hepatocytes’ nucleus, which is responsible for the maturation of hepatocytes, forming cells that are cuboidal in shape expressing the functional genes [67].Regarding the postnatal growth of the liver,β-catenin knockdown in mice proved that it is involved essentially in the hepatocyte proliferation, which is synchronized with the liver size, with the decrease in the liver weight to body weight ratio in the knockout mice signifying the same [68].Wnt/β-catenin involvement in the regeneration of the adult liver is demonstrated by the 2.5-fold increase inβ-catenin and its nuclear translocation within a few minutes in a partial hepatectomy model [69].β-catenin is an important regulator since it activates the oval cells and the bipotential progenitor cells [70].The long-known fact of hepatic zonation into periportal and perivenous zones is also dictated by Wnt/β-catenin pathway.Periportal zone is characterized by higher expression of APC and subsequently lower expression ofβ-catenin and Wnt-targeted genes.Ironically, in the perivenous zone, the expression of APC andβ-catenin isviceversa[68].Hepatocyte nuclear factor-4α(HNF-4α), a transcription factor in the liver, is responsible for the expression of periportal genes in hepatocytes, suppressing theβ-catenin and TCF4 dependent gene transcription and the later suppressing HNF-4αdependent transcription.This is also an important baseline for hepatic zonation [71].β-catenin also regulates the metabolism of ammonia and glutamine in specific hepatic zones.Glutamine metabolism, regulated by three different genes encoding glutamine synthetase, ornithine aminotransferase, and the glutamate transporter GLT-1, is also controlled by Wnt/β-catenin signaling [72].For ammonia metabolism or detoxification, abnormal activation ofβ-catenin results in increased expression of glutamine synthetase in the periportal zone, which is a normal behavior in the perivenous zone.In addition, there is a decreased expression of a urea synthetic enzyme, carbamoyl phosphate synthase, in the periportal zone.Similarly, glutamine synthetase expression is reduced in the perivenous zone when there is a loss ofβ-catenin that leads to elevated plasma ammonia levels [73].Wnt/β-catenin signaling also regulates bile acid secretion.Liver-specificβ-catenin knockout mice were used to find the direct influence on the rate of bile acid secretion and development of intrahepatic cholestasis in the absence ofβ-catenin [74].The role ofβ-catenin in bile canalicular morphogenesis is also proven.β-catenin knockout mice possessed dilated, tortuous bile canaliculi with a paucity of microvilli on the canalicular surface of hepatocytes [75].
Modulation of Wnt/ β-catenin pathway during lipogenesis in NAFLD
Dysregulation of Wnt/β-catenin signaling and its various components affects NAFLD, beginning from early obesity, diabetes,NASH, progressing to fibrosis and HCC.The two different Wnt signaling, namely, canonical and non-canonical pathways, have their own influence on NAFLD.The non-canonical pathway is involved in the furtherance of fat accumulation, inflammation, lipid accretion, promoting NASH.Whereas the classical/canonical pathway involvingβ-catenin functions as an anti-inflammatory, anti-lipid accretion signaling, also mainly responsible for adipocyte differentiation [76].Hence, inhibition or downregulation of the classical Wnt/β-catenin pathway contributes to the onset and progression of NAFLD.However, NAFLD can be inhibited by the upregulation of peroxisome proliferator-activated receptorγ(PPAR-γ), which is a downstream target of Wnt/β-catenin signaling that promotes preadipocytes differentiation, adipogenesis, absorption of FFA and even suppresses inflammation [77].Conversely, sirtuin 1 (SIRT1)was found to activate Wnt/β-catenin pathway by deacetylation of the histones of sFRPs and deacetylation ofβ-catenin, which prevented adipogenesis and consequently fat deposit [78].
Mutations inLRP6are one of the major causes of NAFLD induction.LRP6knockout mice (LRP6+/- mice) fed with a high-fat diet(HFD) were protected against insulin resistance and fat-induced obesity.Body weight and total body fat were significantly decreased in LRP6+/- mice than in the wild-type, denoting that LRP6 causes insulin resistance and increased adiposity, leading to obesity [79].MutatedLRP6(LRP6R611C), arginine to a cysteine residue at 611, results in impaired LRP phosphorylation in Wnt signaling causing the inhibition ofβ-catenin and TCF nuclear translocation.This hinderance is involved in elevated lipid accumulation via different signaling pathways, including insulin-like growth factor 1 (IGF-1)/Akt/mammalian target of rapamycin (mTOR)/sterol regulatory element-binding transcription factor (SREBP)1/2 [80].Deprived levels of Wnt1 and higher levels of DKK-1, an inhibitor of the Wnt pathway in plasma, are correlated with a raised risk of hyperlipidemia by antagonizing LRP6 [81].Wnt signaling is an essential regulator of hepatic metabolism in mammals.Apart from glucose and glycogen metabolism, lipid and TG metabolism are also influenced byβ-catenin.Under the oxidative stress condition,βcatenin binds to forkhead box protein O (FoxO) transcription factors controlling lipid accumulation.Overexpression ofβ-catenin in the hepatocytes of mice results in increased nuclear translocation of FoxO1 [82].The networking between FoxO1 regulation by its phosphorylation, via stimulation of Akt pathway by insulin sensitization [83]is also impacted byβ-catenin.β-catenin deleted mice showed increased insulin-dependent-Akt and GSK-3βphosphorylation, leading to insulin tolerance.Contrastingly,β-catenin overexpression hinders insulin signaling and declines glycogen synthesis [82].Transcription factor variant, transcription factor 7 like-2(TCF7L2), a Wnt signaling component, directly downregulates the standard insulin secretion and confers risk of type 2 diabetes mellitus [84]contributing to the development of NAFLD.Inhibition of DKK-2 improves glucose tolerance in type 2 diabetic mice [85].Mutations producing inactivated LRP6 in mice are capable of instigating lipid accumulation, steatohepatitis, and fibrosis [86].These studies have clearly confirmed that the downregulation of Wnt signaling regulators induces the progression of NAFLD.
Ironically, upregulation ofβ-catenin signaling can suppress NAFLD progression.For instance, liver-specificβ-catenin knockout mice (Ctnnb1 loxp/loxp ; Albumin-Cre-/ + or Ctnnb1 loxp/loxp ; Afp-Alb-Cre-/+) developed severe steatohepatitis when fed with methionine and choline-deficient diet with almost 80% to 100% of the liver parenchyma cells exhibiting steatosis, significant inflammation and hepatocyte ballooning.Knockout mice also manifested a high level of alanine aminotransferase, indicating severe liver injury.A higher degree of TG accumulation is also significantly maintained [87].Elevated interaction betweenβ-catenin and TCF results in downregulation of genes encoding glucose-6-phosphatase and phosphoenolpyruvate carboxykinase, the rate-limiting enzymes in hepatic gluconeogenesis.In contrast,β-catenin and FoxO1 interaction upregulates the above-mentioned enzymes contributing to hyperglycemic condition [60].Apart from the canonical Wnt pathway,the non-canonical Wnt signaling involving Wnt5a, is responsible for the progression of NAFLD to hepatic steatosis.NASH is triggered by Wnt5a via activation of JNK signaling consequently provoking insulin resistance [88].This was proven in a study using a mice model wherein decreased levels of hepatic TGs were observed when the Wnt5a-mediated JNK pathway was inhibited by an endogenous Wnt5 suppressor, sFRP5 [89].These findings signify thatβ-catenin is of major importance in NAFLD and especially its interaction with different downstream Wnt signaling regulators determine the progression of NAFLD.The canonical and noncanonical Wnt signaling pathways in hepatocytes are presented in Fig.2.
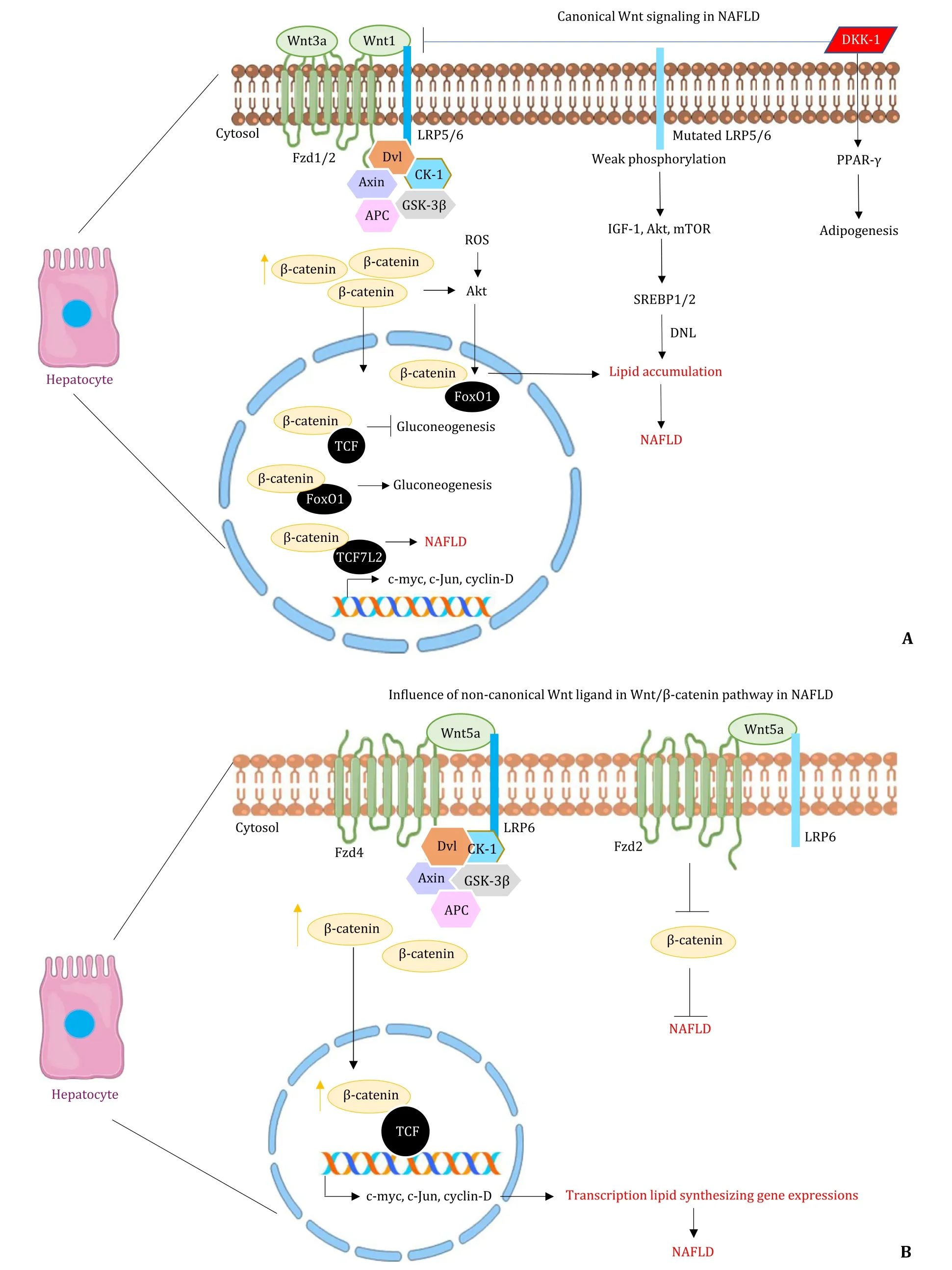
Fig.2.Wnt signaling in hepatocytes during NAFLD.A: The picture shows the activation of Wnt signaling by various canonical Wnt ligands, resulting in higher β-catenin levels and the mutated LRP6 activating pathways like IGF-1, Akt, mTOR.These lead to the expression of genes involved in lipid accumulation in hepatocytes.B: The picture shows the involvement of a non-canonical Wnt ligand (Wnt5a) in the Wnt/ β-catenin pathway in NAFLD.NAFLD is progressed or suppressed depending upon the binding of Wnt5a to different receptors, Fzd4 and Fzd2, respectively.NAFLD: nonalcoholic fatty liver diseases; Fzd: Frizzled receptor; Dvl: Dishevelled; CK-1: casein kinase-1; APC:adenomatous polyposis coli; GSK-3 β: glycogen synthase kinase 3 beta; FoxO1: forkhead box protein O 1; TCF: T-cell factor; ROS: reactive oxygen species; IGF-1: insulinlike growth factor; mTOR: mammalian target of rapamycin; SREBP: sterol regulatory element-binding transcription factor; DNL: de novo lipogenesis; PPAR- γ: peroxisome proliferator-activated receptor- γ; DKK-1: Dickkopf; LRP6: low-density lipoprotein receptor-related protein 6.
Modulation of Wnt/ β-catenin pathway during NAFLD mediated fibrosis
Progression of NAFLD to fibrosis is one of the major attributes observed in patients.The Wnt/β-catenin pathway also influences the process of fibrosis.Genomic analysis of hepatic fibrosis in primary biliary cirrhosis liver revealed the elevated expression of Wnt13, Wnt5a, andβ-catenin [90].Various components of Wnt/βcatenin signaling like Wnt3a, Wnt10b, Fzd receptor 1, 2, LRP6,βcatenin and TCF have proven to be critical in the onset and progression of fibrosis.These are involved in HSC activation and consequent expression of these factors in activated HSCs [91].Wnt3a plays a predominant role in activating human HSCs, simultaneously stimulating collagen I and III production, while also preventing the TNF-related apoptosis-inducing ligand (TRAIL)-induced apoptosis of HSCs.Wnt3a is also negatively regulated in cells overexpressing sFRP1 and DKK-1 (Wnt signaling antagonists), which induce apoptosis in cultured HSCs [ 91 , 92 ].Wnt5a is another inducer of fibrosis.Inhibition of Wnt5a reduces the activation of HSCs [93].The documentations show that activation, inhibition, and even noninvolvement ofβ-catenin promote Wnt5a activity in hepatic fibrosis.Similarly, the opposing and favoring effect of Wnt5a onβ-catenin activity depends on the Fzd receptor types.Whether Wnt5a activates or inhibitsβ-catenin signaling depends on the type of Fzd expressed by a cell.Wnt5a activatesβ-catenin signaling in the presence of Fzd4, and inhibitsβ-catenin in the presence of Fzd2 [60].A study involving rat HSCs cell line proved that siRNA mediated knockout ofβ-catenin resulted in the inhibition of HSC proliferation, an increase in apoptosis and especially inhibition of collagen I and III synthesis [94], suggesting thatβ-catenin siRNA could be useful to regress hepatic fibrosis by controlling HSCs activation.Mesoderm-specific transcript homologue in HSCs downregulatedβ-catenin expression, thus suppressing CCl 4 -induced collagen accumulation bothinvitroandinvivo,and also the viability of HSCs [95].According to the study, Wnt1 or mutated S33Yβ-catenin in preadipocytes is a negative regulator of adipogenesis at least partially by inhibition of PPAR-γ, which might activate the HSCs and fibrosis.Invitroandinvivomodels exhibited myofibroblasts like cells without differentiating into adipocytes, signifying the role of the Wnt pathway in HSCs activation ( Fig.3 ) [96].Inhibition of Wnt signaling substantiated the adipogenic gene profile of quiescent HSC, preventing fibrosis.DKK-1 (a Wnt signaling antagonist) was found to be present in both quiescent and activated HSCs [91].But DKK-1 played its role in the maintenance of HSC quiescence by increasing PPAR-γexpression, consequently inducing adipogenesis [ 97 , 98 ].However, DKK-1 suppressed hepatic fibrosis by inhibiting the fibrogenic markers likeα-SMA, type I collagen and TGF-β1 on activated HSCs in animal models [91].In contrast, significant changes were not observed between the quiescent and the activated HSCs in the phosphorylation and nuclear translocation ofβ-catenin in the DNA microarray studies [99].These data collectively prove that the upregulation of Wnt signaling by various Wnt/β-catenin regulators, its interaction and the involvement of non-canonical Wnt ligand are crucial in the development of fibrosis.
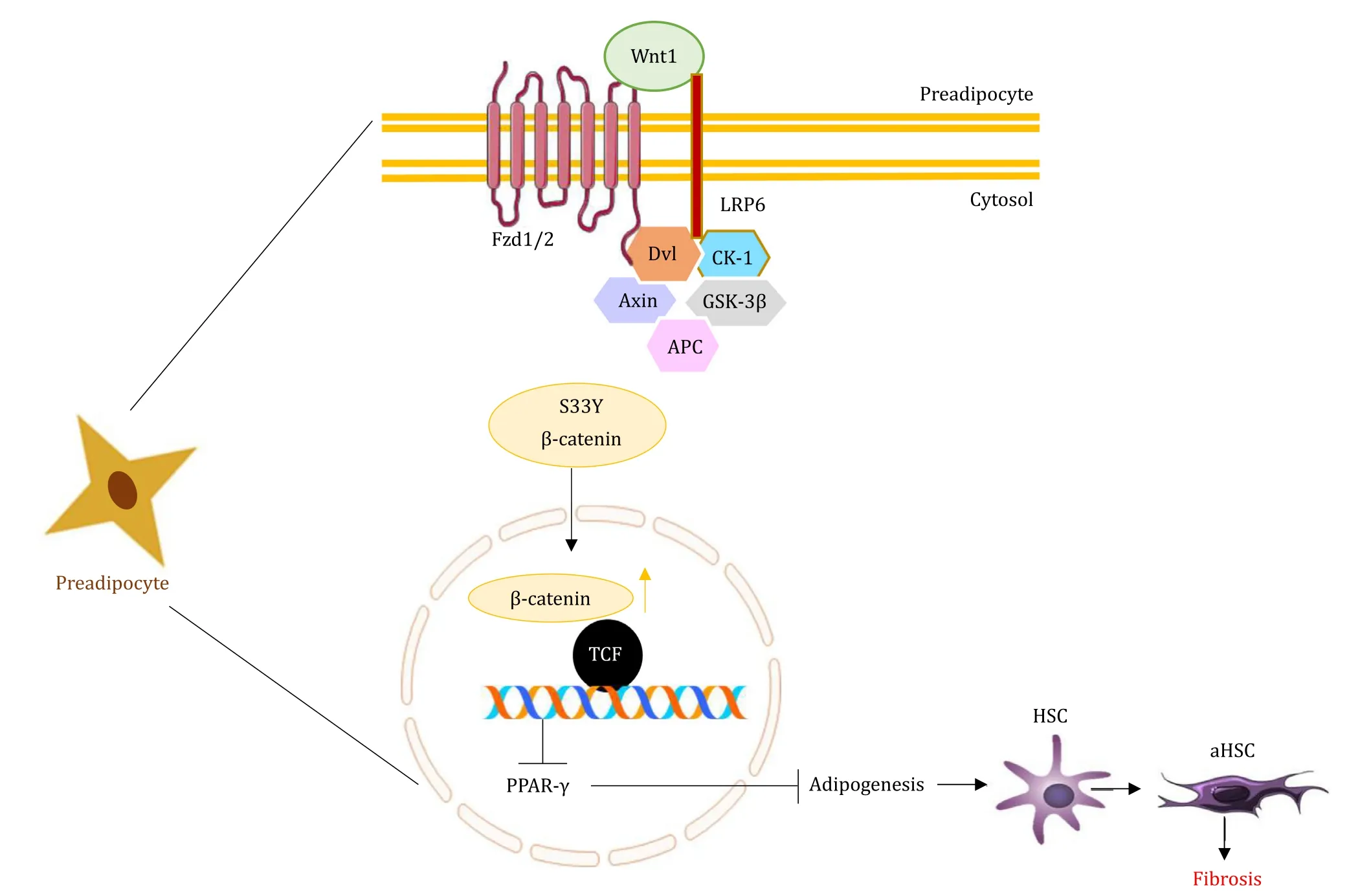
Fig.3.Wnt signaling in preadipocytes in NAFLD.The picture shows the activation of Wnt signaling by Wnt1 that suppresses PPAR- γ expression and adipogenesis which stimulates the activated HSCs resulting in fibrosis.NAFLD: nonalcoholic fatty liver diseases; Fzd: Frizzled receptor; LRP6: low-density lipoprotein receptor-related protein 6;Dvl: Dishevelled; CK-1: casein kinase-1; APC: adenomatous polyposis coli; GSK-3 β: glycogen synthase kinase 3 beta; TCF: T-cell factor; PPAR- γ: peroxisome proliferatoractivated receptor- γ; HSC: activated hepatic stellate cell.
Increased ROS production and oxidative stress has been implicated in the NAFLD progression via Wnt/β-catenin signaling [100].ROS stimulates Wnt ligands, Fzd receptors in HSCs and Kupffer cells and results in fibrosis.Experimentation with male C57BL/6J mice fed with methionine and a choline-deficient diet proved that the upregulated expression of Wnt1a was associated with ROS production and that its downstream elements were responsible for fibrogenesis [101].Various Wnts and Fzds were observed in quiescent and activated HSCs and Kupffer cells in the liver.Fifteen Wnts (Wnt2, Wnt2b, Wnt3, Wnt4, Wnt5a, Wnt5b, Wnt6, Wnt7a,Wnt8b, Wnt9a, Wnt9b, Wnt10a, Wnt10b, Wnt11, and Wnt16) and seven Frizzled receptors (Fz2, Fz3, Fz4, Fz6, Fz7, Fz8, and Fz9) were present in both resting and active cells [102].However, it was found that the ‘Fzb’ receptor type was expressed only in activated HSCs and Kupffer cells.Wnt signaling also influences the liver sinusoidal endothelial cells in terms of fibrosis.Overexpression of Wnt2 and Fzd4 and translocation ofβ-catenin in the liver sinusoidal endothelial cells reveal the effect of Wnt signaling on fibrosis [103]( Fig.4 ).
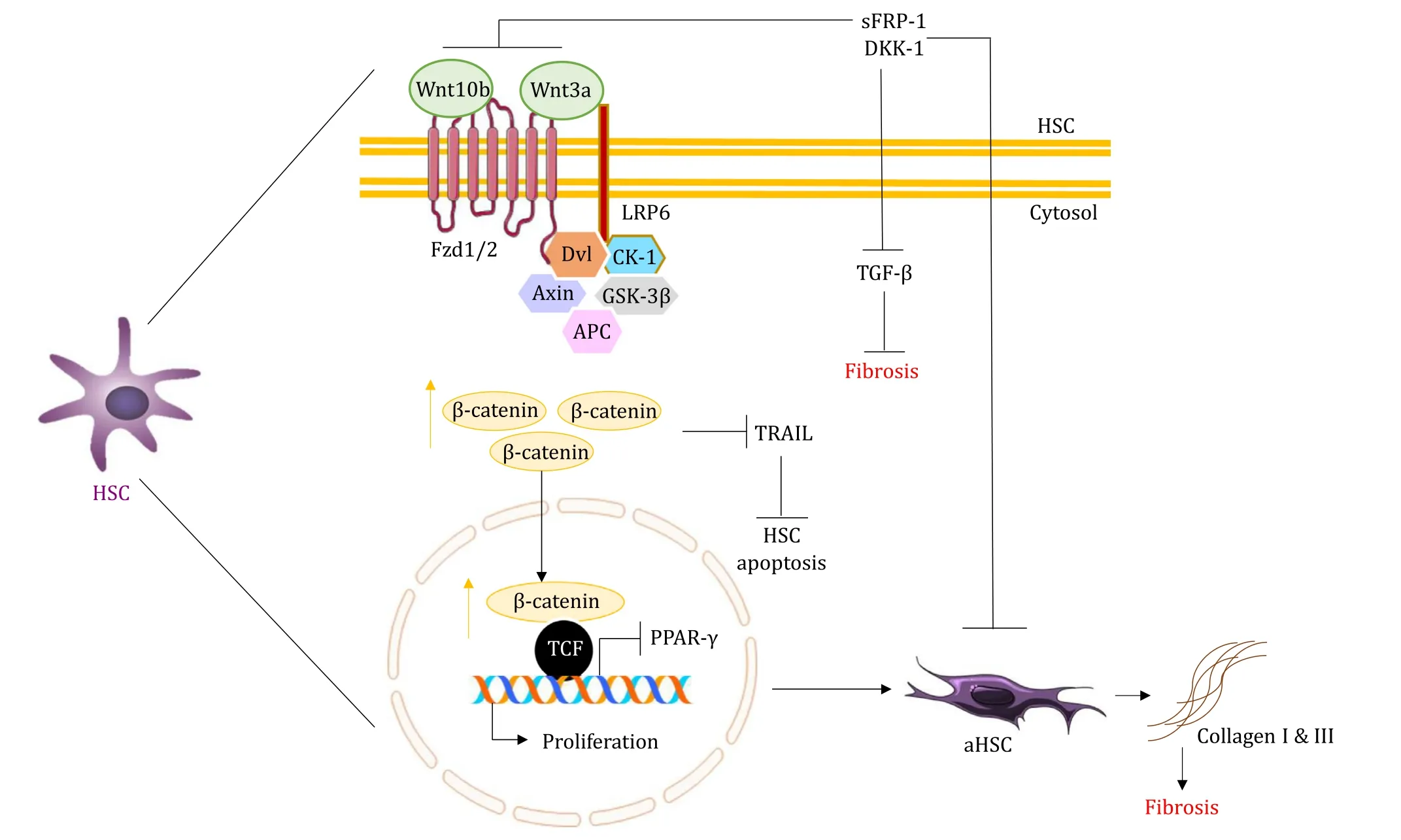
Fig.4.Wnt signaling in HSCs in NAFLD.The picture shows the role of Wnt/ β-catenin signaling in the activation of genes responsible for the proliferation and activation of HSCs that produce extracellular matrix (ECM) like collagen, resulting in fibrosis.NAFLD: nonalcoholic fatty liver diseases; β-catenin also promotes NAFLD by preventing HSC apoptosis via suppression of tumor necrosis factor-related apoptosis-inducing ligand (TRAIL).This Wnt signaling can be inhibited by Dickkopf (DKK-1) and secreted Frizzledrelated proteins (sFRP-1), preventing fibrosis.NAFLD: nonalcoholic fatty liver diseases; Fzd: Frizzled receptor; LRP6: low-density lipoprotein receptor-related protein 6; Dvl:Dishevelled; CK-1: casein kinase-1; APC: adenomatous polyposis coli; GSK-3 β: glycogen synthase kinase 3 beta; TCF: T-cell factor; PPAR- γ: peroxisome proliferator-activated receptor- γ; TGF- β: transforming growth factor- β; aHSC: activated hepatic stellate cell.
The complexity of Wnt signaling in NAFLD is demonstrated by various studies involving the role of the classical Wnt pathway in preventing its progression.For instance, a study demonstrated that activation of Wnt/β-catenin signaling can ameliorate liver injury by restoring the HPCs via the activation of SIRT1 [104].However,over activation of classical Wnt pathway components like Wnt3a in macrophages stimulatedβ-catenin phosphorylation in HPCs in a pediatric NAFLD study [105].Further intense in the NAFLD, the disease progresses and leads to the development of HCC, which is promoted by impaired Wnt signaling.Mutations in theCTNNB1gene encodingβ-catenin, APC, and AXIN1/2 result in the persistent stabilization ofβ-catenin and expression of Wnt target genes [106].Significantly higher expression of LRP6 mRNA was reported in HCC cell lines (Hep3B and Huh-7).LRP6 knockdown in Huh-7 cells minimized theβ-catenin pool and TCF/LEF activity, whereas the overexpression of LRP6 restored the activity.These findings were also confirmed by a study on Huh-7 cells, Huh-7 xenograft mice and human clinical samples [107].Various epigenetic modifications in Wnt signaling also result in HCC.DNA methylation of Wnt antagonists like SFRP1,SFRP2,SFRP4 and SFRP5, SRY-box containing gene(SOX17) promotes classical Wnt signaling involved in tumorigenesis [ 108 , 109 ].Histone modifications mediated by enhancer of zeste homolog 2 (EZH2) suppress Wnt signaling antagonists promoting cellular proliferation contributing to HCC [110].Histone deacetylase 8 (HDAC8), belonging to class I HDAC, interacts with EZH2 and stimulates insulin resistance and Wnt/β-catenin pathway in a high dietary NAFLD-HCC mouse model [111].Several miRNAs contribute to the development of HCC via upregulated Wnt signaling.Loss of miRNA-122 in mice models promoted the overexpression of the Wnt pathway, progressing from steatohepatitis to fibrosis to HCC [112].Increased serum level of miRNA-34a is observed due to over activation of the Wnt/β-catenin pathway in mouse tumors and HCC patients [113].Liver cancer cells were reported to be suppressed by SIRT1 via the inhibition of Wnt/β-catenin pathway by phosphorylatingβ-catenin in protein kinaseα-dependent manner [114].Contrastingly, SIRT1 inhibitor suppressed cell migration facilitated by Wnt/β-catenin pathway.Hence, we can conclude that the overexpression of Wnt/β-catenin pathway by several mechanisms has a prominent effect in the development of advanced stages of NAFLD.
Wnt/ β-catenin pathway modulators
The data collected and the studies performed so far denote that the demand for an effective treatment of NAFLD is reaching its heights and that Wnt signaling seems to be an effective target.Thus, with all the knowledge gathered on the structure, mechanism, and function of Wnt signaling and its components, various studies have so far been conducted on its modulators to prevent diseases and restore the normal functionality of organs.Compounds ranging from natural extracts to chemically synthesized molecules and endogenously produced components prove to be effective in modulating or blocking the Wnt/β-catenin pathway.Several drugs and small molecules have been identified to target Wnt signaling in different steps and its constituents [115].Natural compounds like chalcones including derricin effectively reducedβ-catenin in the nucleus preventing its interaction with DNA [116].Carnosic acid extracted from the plant rosemary aids in the reduction of transcriptionalβ-catenin by interfering with B-cell lymphoma (BCL-9) [117].OMP-18R5, an Fzd antibody, blocks Wnt ligands via steric hindrance by binding to Fzd cysteine-rich domains (CRD) resulting in proteasomal degradation ofβ-catenin.OMP-18R5 itself is effective and acts synergistically with taxol, a chemotherapeutic agent used in the treatment of cancer [118].sFRPs with an N-terminal cysteine-rich domains as mentioned earlier as an antagonist of Wnt, bind either to the Wnt ligand or to the Fzd receptor, thereby blocking its ability to bind to Wnt protein [119].Sclerostin binds to the first or to the first and second domains of LRP5/6, inhibiting the binding of Wnt, obstructing the canonical Wnt pathway [120].NSC668036 and 3289-8625 are small molecules that disrupt the interaction between Fzd and PDZ domain of Dvl protein which is quite essential for the downstream transduction of Wnt/β-catenin signaling [ 121 , 122 ].Further,insilicostudies proved that J01-017a is the most potent Dvl-PDZ domain inhibitor [123].Sulindac, a nonsteroidal anti-inflammatory drug binds to the PDZ domain of Dvl with the specificity to the side chain in arginine residue 322.This binding subsequently quenches the Wnt/β-catenin signaling components.Studies on sulindac proved it to be a good bioavailable compound with the ability to cross the blood-brain barrier, significantly reducing tumor growth by inducing apoptosis and without being toxic to the non-tumor environment [ 124 , 125 ].These are the small molecules and drugs tested against Wnt/β-catenin pathway and were proven to be effective against various diseases, including cancer.
Synthetic molecules and drugs modulating Wnt/ β-catenin signaling in NAFLD
Drugs have been developed specifically to act against the Wnt/β-catenin signaling in NAFLD.These include small molecules,synthetic compounds and drugs that exert their action on various components of Wnt signaling ( Table 1 ).An FDA-approved antipinworm drug, pyrvinium, is now regarded as a Wnt/β-catenin inhibitor by decreasing theβ-catenin levels and its downstream constituents in a time and dose-dependent manner [126].Pyrvinium,a highly potent small-molecule, binds to casein kinase 1α, leading to the degradation ofβ-catenin, halting Wnt signaling [127].Pyrvinium pamoate significantly reduced the HFD-induced hepatic lipid accumulation, insulin resistance, oxidative stress, hepatic inflammation and fibrosis in rat by inhibiting the Wnt/βcatenin pathway.Pyrvinium pamoate treatment was quite successful in reducing c-myc, Fzd7, Wnt3a andβ-catenin gene expression [128].Celecoxib decreased the serum glucose, TG, cholesterol level, Wnt5a, JNK1 p54 and JNK1 p46 expressions and promoted insulin sensitivity in type 2 diabetes mellitus-NASH rats [129].Injured hepatocytes respond by stimulating the overexpression of A3 adenosine receptor (A3AR) in the inflammatory cells in liver, which secrete proinflammatory molecules.Namodenoson(2-chloro-N6-(3-iodobenzyl)-adenosine-5’-N-methyl-uronamide), a synthetic adenosine derivate, binds specifically to A3AR and exerts its anti-inflammatory effects on injured hepatocytes in xenograft liver cancer models [130].Namodenoson is also an anti-steatotic,anti-fibrotic, and anti-tumorigenic drug.A study involving phase I and II trials established that HCC is suppressed by namodenoson that induces apoptosis of HCC cells by dysregulating Wnt/βcatenin signaling [131].Namodenoson further upregulated GSK-3βand downregulatedβ-catenin, Lef-1 and cyclin D1 in C57BL/6 mice, CCl4-induced fibrotic mice and LX2 cells [132].Pimozide, an anti-psychotic drug, is an FDA-approved drug recently noted for its ability to inhibit epithelial cell adhesion molecule, which is a downstream component of the Wnt pathway.Pimozide inhibits HCC cellular proliferation in a dose-dependent manner, and reduces the expression ofβ-catenin, cyclin D1 in Hep3B and HepG2 cells [133].Itraconazole, an FDA-approved antifungal drug is now found to possess antitumor activity [134].Itraconazole at a concentration of 2 and 8μg/mL resulted in the decrease of Wnt3a with the increase in its inhibitor, Axin-1.Significant reduction in theβ-catenin levels is also an attribute of itraconazole in HepG2 and Bel-7405 cell lines [135].Contradicting to the mechanism of Wnt/β-catenin signaling suppression to treat NAFLD, upregulation of Wnt3a in Kupffer cells is involved in the suppression of NAFLD.Docosahexaenoic acid possesses an anti-inflammatory effect on macrophages [136].And its treatment increases Wnt3a+macrophages, increases anti-inflammatory macrophages (CD206+)and decreases the inflammatory (S100A9+) macrophages.Docosahexaenoic acid is also found to induce the phosphorylation ofβcatenin in HPC, leading to its proteasomal degradation.All of these significantly reduce steatosis and proinflammatory cytokines in pediatric NAFLD patients [105].
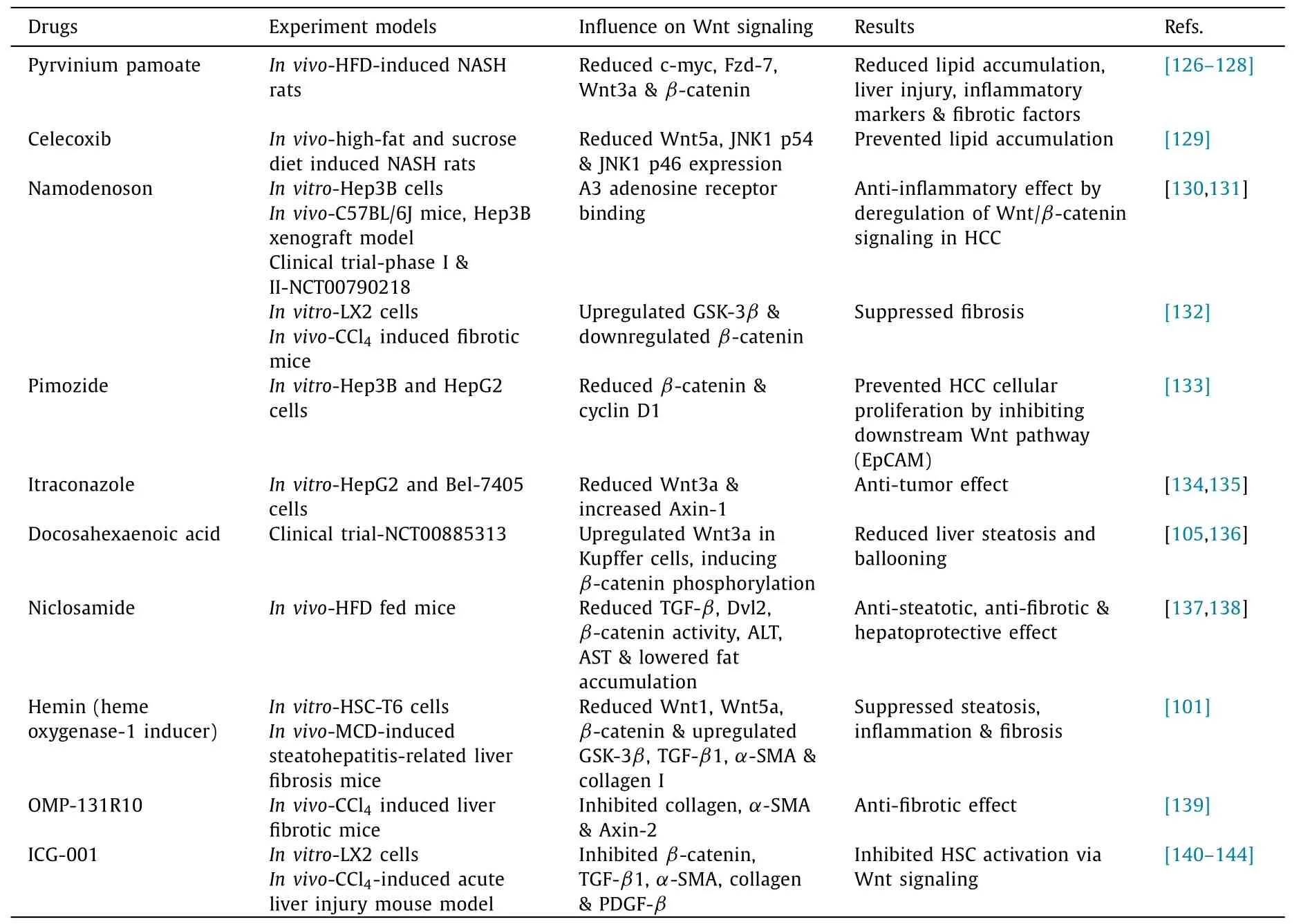
Table 1Small molecules modulating Wnt/ β-catenin signaling in NAFLD.
In HFD fed mice, an anti-helminthic drug, niclosamide, administered as niclosamide ethanolamine salt, decreased body weight,diabetes, and consequently reduced steatosis by lowering fat accumulation [137].Oral administration of niclosamide sufficiently ameliorated hepatic fibrosis by reducing serum aminotransferases,TGF-β, Dvl2, andβ-catenin activity [138].Hemin is an inducer of heme oxygenase-1 that plays a significant role in the treatment of steatohepatitis-related hepatic fibrosis.Hemin induced heme oxygenase-1 proved to significantly upregulate the expression of hepatic GSK-3β.It also suppressed Wnt1, Wnt5a,β-catenin, TGFβ1,α-SMA and collagen I expression in methionine and cholinedeficient diet-induced steatohepatitis-related hepatic fibrosis mice and in HSC-T6 cells [101].OMP-131R10, an anti-RSPO3 antibody(R-spondin 3, a Wnt/β-catenin amplifier), inhibits collagen synthesis andα-SMA expression in CCl 4 -induced hepatic fibrosis in mice.It also efficiently inhibits the expression of a Wnt target gene,Axin2[139].Upstream regulators ofβ-catenin include vantictumab [anti-Fzd1/2/5/7/8 monoclonal antibody (aAb)], IgG-2919 (anti-Fzd5 mAb), rosmantuzumab (anti-RSPO3 mAb), ipafricept (PORCN inhibitors: Fzd8-Fc, ETC-159, GNF6231, WntC59 and Wnt974), AZ1366, G007-LK, NVP-TNKS656 and XAV939 (TNKS inhibitors) [ 118 , 140 , 141 ].The interaction between TCF andβcatenin is inhibited by molecules like CGP049090, CWP232228 and LF3 [ 142 , 143 ].These drugs have anti-fibrotic and anti-tumor effects.ICG-001, a Wnt/β-catenin/CBP inhibitor, exerts dosedependent inhibition ofβ-catenin, TGF-βinduced collagen I,α-SMA, PDGF-βand TIMP with the reduction in total liver collagen content in CCl4-induced acute liver injured mice.ICG-001 also proved to attenuate HSC activation via inhibiting Wnt signaling in LX2 cells [144].
Herbal based Wnt/ β-catenin signaling modulators in NAFLD
Apart from the synthetic molecules, there are herbal extracts and medicines which are also effective against NAFLD( Table 2 ).Isoflavones inhibit Wnt5a expression in adipose tissue and adipogenesis-related genes.For instance, genistein, an isoflavone, suppresses adipocyte differentiation by activating the Wnt pathway via upregulation of Wnt3a andβ-catenin and downregulating the antagonists of Wnt signaling like DKK2,SFRP1 [145].Morin is a bioflavonoid compound known for its antitumor, anti-inflammatory and anti-oxidant properties [146-148].Morin suppressed the key regulators of Wnt signaling such as GSK-3β,β-catenin, cyclin D1 and c-myc, Wnt3a and Wnt5a in LX-2 cells [149].Progression of NAFLD to hepatic carcinogenesis can also be treated by targeting Wnt signaling.S-allylmercaptocysteine extracted from garlic directly interconnects with LRP6 on the cancer cell membrane to block Wnt signaling, thereby promoting apoptosis of HCC cells via c-myc [107].Tetrandrine, a herbal compound obtained from a Chinese medicinal herbStephaniatetrandra[150],suppresses the migration and metastasis of HCC by downregulating the nuclear translocation ofβ-catenin via tetrandrine-induced autophagy [151].
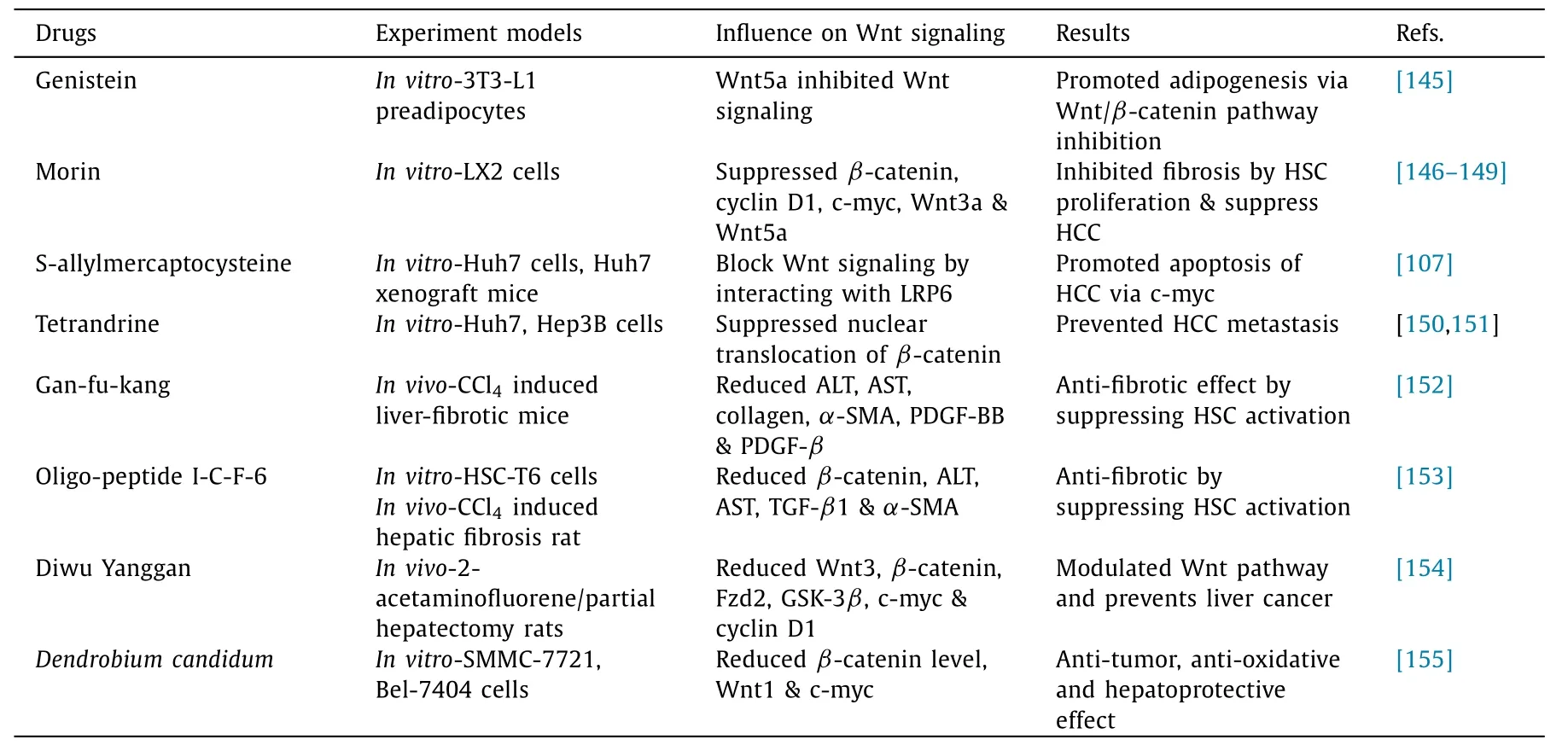
Table 2Herbal based Wnt/ β-catenin signaling modulators in NAFLD.
Gan-fu-kang, a traditional Chinese herbal cocktail containingSalviamiltiorrhiza,Astragalusmembranaceus,Radixpaeoniaealba,etc., is used as a hepatoprotective and anti-fibrotic drug.Ganfu-kang significantly downregulated Wnt signaling and inhibited HSC activation in CCl 4 -induced hepatic fibrotic mice [152].Another Chinese traditional medicine,Carapaxtrionycis,has a component oligo-peptide I-C-F-6 that suppresses hepatic fibrosis-inhibiting Wnt/β-catenin signaling.The time and dose-dependent effect of oligo-peptide I-C-F-6 was observed as a decrease inβ-catenin levels that eventually mitigates HSC-T6 proliferation, and TGFβ1 levels, thereby decreasing collagen 4, and TIMP-1 expression [153].Diwu Yanggan, a Chinese herbal medicine containing a combination of plant extracts, was shown to reduce the expression of Wnt3,β-catenin, Fzd2, GSK-3β, c-myc and cyclin D1 in 2-acetaminofluorene/partial hepatectomy animal models [154].Dendrobiumcandidum, a Chinese herbal plant and its components like polysaccharides, aromatic compounds and sesquiterpenoids exert antioxidant, anti-tumor and hepatoprotective activity.Dendrobiumcandidumextract decreased the nuclearβcatenin level, Wnt1 and c-myc expressions in SMMC-7721 and Bel-7404 liver cancer cell lines [155].Since herbal medicines are known for their less or no side effects in treating diseases, these studies prove to be of great importance in the treatment of NAFLD.
Future perspectives
The escalating number of NAFLD cases among the population, including adults, adolescents, and children, and the prediction of its increased burden in the next two decades demand us to look for effective means of treatment.As complex as the disease is, its mode of progression and its association with other organs and disorders, targeting Wnt/β-catenin proves to be a potent treatment.Previously established Wnt/β-catenin signaling modulators like derricin, sclerostin, small molecules like NSC668036, 3289-8625 and J01-017a can be tested further for its role against NAFLD particularly.Since there are so many target points in the Wnt signaling itself, studying and uncovering the best site of action would be helpful.Drugs including pyrvinium pamoate, pimozide, niclosamide, itraconazole, and ICG-001 have been studied against some of the stages of NAFLD.Their antisteatotic, anti-inflammatory, anti-fibrotic, and anti-tumor effects wholly against NAFLD need further studies.Also, with the advancement of so many drug discoveries, the most potent drug at minimal dosage without or with less side effects to target Wnt signaling and also its combinatorial effect to treat NAFLD can be studied.Furthermore, expanding the studies on drugs that can treat NAFLD along with its various other associated diseases will set a benchmark.
Conclusions
NAFLD is one of the most common chronic liver diseases.It is characterized by the hepatic accumulation of excess fat in people who consume little or no alcohol.NAFLD is a worldwide disease in people of different ages.The progression of NAFLD is complex,beginning with simple fat accumulation that intensifies into major conditions like NASH, fibrosis, and HCC.The participation of Wnt/β-catenin is essential.Various components of the Wnt signaling pathway, including LRP6, Wnt1, Wnt3a,β-catenin, GSK-3β, and APC, are regulated in such a way that promotes NAFLD progression.The Wnt signaling cross talks with other pathways, like TGFβ, mTOR, and PK13 aggravates the disease.Such Wnt signaling and its components are targeted effectively by various synthetic drugs,herbal compounds, and endogenously available inhibitors.Collectively, all the previously identified drugs exert its inhibition mechanism on Wnt signaling at specific step, at appropriate concentrations, thus suppressing the NAFLD pathogenesis.
Acknowledgments
None.
CRediT authorship contribution statement
Karthik Shree Harini:Writing - original draft, Writing - review & editing.Devaraj Ezhilarasan:Conceptualization, Formal analysis, Funding acquisition, Project administration, Resources,Software, Validation, Writing - review & editing.
Funding
The study was supported by a grant from the Extramural Research Project, Indian Council of Medical Research, New Delhi, India (58/30/2020/PHA/BMS dtd 9.11.2021).
Ethical approval
Not needed.
Competing interest
No benefits in any form have been received or will be received from a commercial party related directly or indirectly to the subject of this article.
 Hepatobiliary & Pancreatic Diseases International2023年4期
Hepatobiliary & Pancreatic Diseases International2023年4期
- Hepatobiliary & Pancreatic Diseases International的其它文章
- Hypermethylation of thymosin β4 predicts a poor prognosis for patients with acute-on-chronic hepatitis B liver failure
- Novel re-intervention device for occluded multiple uncovered self-expandable metal stent (with video)
- The impact of metabolic dysfunction-associated fatty liver disease on the prognosis of patients with hepatocellular carcinoma after radical resection
- Laparoscopic liver resection for hepatocellular carcinoma complicated with significant portal hypertension: A propensity score-matched survival analysis
- Hepatobiliary&Pancreatic Diseases International
- Radiomics in the diagnosis and treatment of hepatocellular carcinoma