
药物-药物共晶的设计与制备方法及应用研究进展*
2023-07-05 14:14王烨阳袁鹏辉杨德智邢逞张丽吕扬
医药导报 2023年7期
王烨阳,袁鹏辉,杨德智,邢逞,张丽,2,吕扬
(1.北京协和医学院、中国医学科学院药物研究所,晶型药物研究北京市重点实验室,北京 100050;2.山东益康药业有限公司,山东省晶型药物重点实验室,滕州 277500)
药物共晶是活性药物成分(active pharmaceutical ingredient,API)和共晶形成物(cocrystal former,CCF)通过非共价相互作用以固定的化学计量比结合在同一晶格的晶体[1]。两种药物活性成分结合在同一晶格中形成的复合物为药物-药物共晶。药物共晶不同于固定剂量组合(fixed dose combination,FDC)药物,后者是指两种及两种以上原料药以固定比例组合成单一剂量形式[2]。传统的FDC药物可减少药品服用数量,简化给药方案,提高患者依从性。此外,FDC策略还可以降低药品生产成本[3]。然而,FDC作为普通物理混合物[4],存在稳定性及溶解度差异、药物间化学兼容性差、生物利用度降低等缺点,而这些恰恰是决定API疗效的关键因素[5]。因此,迫切需要一种新的技术方法,来促进药物-药物混合配方的开发以解决此类问题。药物-药物共晶技术可以在不改变药物化学结构的基础上,改善母体药物的理化性质(如溶解性、稳定性和生物利用度等),发挥协同治疗和减毒增效作用,从而解决常规FDC药物和联合用药治疗中的相关问题[6]。ZHANG等[7]制备了由两种体内溶解性和生物利用度较低的抗血栓药物阿哌沙班和槲皮素组成的新型药物-药物共晶体,与阿哌沙班原料药相比,药物-药物共晶溶解度、生物利用度和稳定性均得到明显改善。此外,药物-药物共晶的新物质状态符合申请发明专利审查要求的新颖性、实用性和创造性[8],可突破原研专利壁垒,有效拓展产品销售周期。因此大大激励了制药企业和研发机构投入研发力量集中研究药物-药物共晶的设计新方法。目前,已上市的药物-药物共晶新药品种数量不多(图1)[9],其中包括2015年获批上市的沙库巴曲-缬沙坦(诺欣妥)和2017年获批上市的盐酸曲马多-塞来昔布(Seglentis),部分品种尚处于临床研发阶段,如沙库巴曲和阿利沙坦水解产物 EX3174 组成的共晶等[10]。笔者在本文综述近年来药物-药物共晶的设计方法、制备技术和应用进展,展望药物-药物共晶研究发展趋势,以期为药物-药物共晶的深入研发提供技术方法参考。
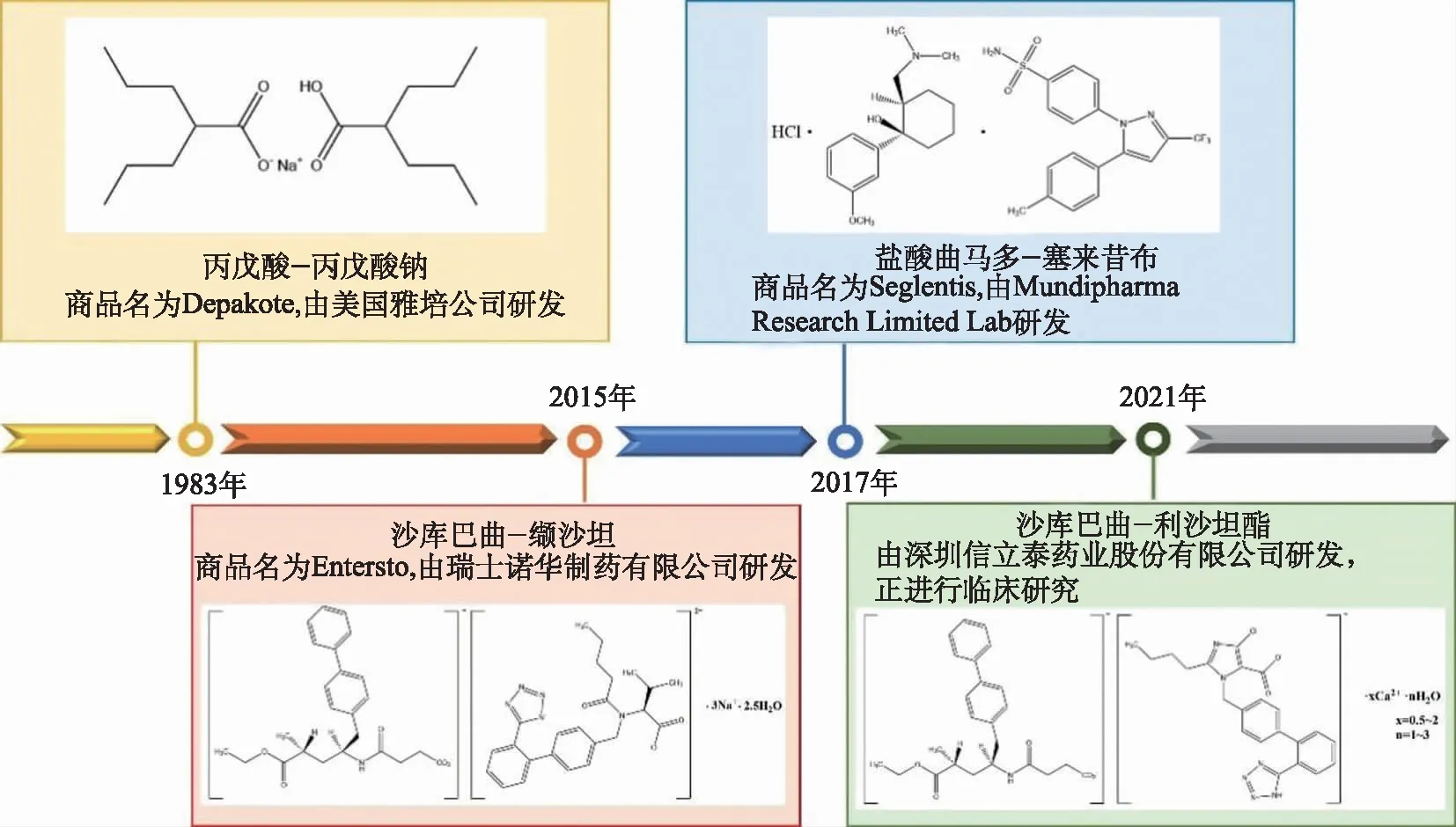
图1 部分已上市或临床研究中的药物-药物共晶品种
1 药物-药物共晶设计预测方法
选择和确定合适的API与CCF是成功开发药物共晶体的关键,也是药物共晶开发的主要难点。对于药物-药物共晶,选择合适的药物组合需要考虑多种因素,不能自由地选择任意两种药物,需要充分考虑不同组分溶解度、分子相互作用、包装模式、配伍剂量、适应证和毒副作用等因素。目前大多数共晶药物筛选通常建立在实验研究的基础上,但这种方法存在代价高昂、耗时耗力,使得许多药物-药物共晶发现具有偶然性,制备效率和速度也存在局限。近年来,随着计算化学和人工智能技术的深入应用,目前已有研究报道将计算化学或人工智能技术方法用于药物-药物共晶设计预测,包括根据解离常数(pKa)规则、汉森溶解度参数进行共晶预测、基于不同类型的“超分子合成子”思路设计共晶、基于云计算晶体结构预测技术、基于分子表面静电势虚拟设计等方法[11-12]。
1.1根据pKa规则预测共晶 根据pKa规则可预测共晶的形成,通过计算两组分间pKa差值大小来衡量两组分之间相互作用。当[ΔpKa(pKa(碱)-pKa(酸)] >3时,发生质子转移,通常形成盐;当0<ΔpKa<3时,质子转移和离子化程度无法明确预测;当ΔpKa<0时,一般不发生质子转移,形成共晶。根据计算获得的ΔpKa(-1.98)<0,选择黄芩素(pKa5.4)和烟酰胺(pKa3.4)成功制备共晶,改善了其溶解性与生物利用度[13]。但该预测方法与实际共晶物的形成可能性间还存在一定差距,通常用于共晶药物的初步预测。
1.2根据汉森溶解度参数进行共晶预测 根据汉森溶解度参数理论,共晶是由API和CCF组成的均相分子混合物,其混相应该与溶解度参数(δ)有关,通过基于组分在固体状态下的混溶性来预测共晶形成的可能性。汉森溶解度参数(t)由分散力(d)、极性力(p)和氢键(h)三个维度的参数构成,可由以下公式确定:
δ2t=(δ2d+δ2p+δ2h)
当CCF溶解度参数更接近API时,容易混合并形成共晶。GHOSH等[14]利用汉森溶解度参数预测烟酸作为CCF可能形成的共晶,制备获得了莫达非尼与烟酸的共晶。该方法需要计算获得溶解度参数,可应用于预测普通药物共晶形成,同时可用于预测药物-药物共晶形成。
1.3基于不同类型的“超分子合成子”思路设计共晶 共晶概念源于超分子化学,大多数API与CCF具有分子间成键基团,其中最常见的是氢键[15]。药物共晶设计本质上是利用API与CCF官能团的互补性设计氢键超分子合成子[16]。通过选用各种筛选方法来确定药物结合位点和CCF之间形成超分子合成的可能性,再合成实验产物并对其进行表征确证。利用超分子原理时,应首先对API的化学结构进行全面分析,包括分子构象、官能团等信息,确定可能的超分子合成子,结合氢键和空间结构的互补性特征,选择合适的CCF。超分子合成子分为两种。①异型合成子:不同但互补官能团之间的合成子,如羧基-酰胺基、羧基-吡啶基、醇羟基-醚基;②同型合成子:相同且互补官能团之间的合成子,如羧基-羧基和酰胺基-酰胺基。通常异型合成子有较强的形成共晶作用[17]。共晶体中存在的超分子合成子的结合方式不同,如图2所示。BOLLA等[18]通过超分子合成子,筛选获得乙酰唑胺与烟酰胺共晶(图2A);LIU等[19]根据异烟肼和吡嗪酰胺具有相似治疗效果且靶点接近的特性,采用一种三元共结晶策略,即药物-桥梁-药物(图2B),在两个API分子之间没有直接接触的情况下,通过一个CCF分子作为桥梁将两个不同的API分子连接到同一个晶格中。插入的第三个CCF分子(桥梁)应该具有两个或更多的氢键位点,这些氢键位点可以与不同API适应匹配,从而形成稳定的超分子合成子。该方法克服了两种API难以直接形成共结晶的问题,为一些具有相似疗效与靶点的药物-药物组合系统的共晶筛选提供了新的设计思路和研究方案。
1.4基于其他物理模型的设计方法 分子表面静电势(molecular electrostatic potential surfaces,MEPs):MEPs通过识别分子表面可能的分子间相互作用位点,根据计算获得氢键参数估算化合物分子相互作用位点配对能,进而预测两种组分形成共晶的概率。该方法计算简单,耗时短,可以用作高通量虚拟筛选工具,但由于忽略了分子构象对MEPs上极值点分布的影响[12],预测结果有时并不理想。YANG等[20]将构象分析引入MEPs计算,分析了3种黄酮类物质山奈酚、槲皮素和杨梅素与吡喹酮的构象,以及不同构象之间可能的相互作用,预测了共晶的形成,并通过实验与各种分析技术结合,验证预测共晶均能形成。随着人工智能技术深入应用并与共晶预测研究结合,越来越多预测方法应用于共晶药物设计[21]。
2 药物-药物共晶制备方法
随着药物晶体学研究的日益深入,目前已有多种不同方法来制备药物-药物共晶体,相关制备方法的优势与不足比较见表1。
2.1固体制备法 固体制备法一般分为干研磨法、加液研磨法和高分子辅助研磨法等。干研磨法是指将两个或多个组分按固定的化学计量比直接混合,手工或机械研磨一段时间;加液研磨法是指在干磨法研磨过程中加入少量溶剂的方法。DA等[22]采用加液研磨法制备了氟尿嘧啶与5-氟胞嘧啶共晶体,与干磨法相比,加液研磨法中由于存在催化量的溶剂,可增强分子识别所需的分子扩散,产生大量非晶相,提高制备效率。高分子辅助研磨法是加液研磨法的替代方法,通过在研磨过程中加入固态或液态聚合物诱导共晶产生。通过研究发现聚合物的加入可增加药物-药物共晶品种的多样性,有效改善共晶的溶出速率和生物利用度[23-24]。
2.2液体制备法 常用液体制备法包括蒸发结晶法、冷却结晶法、喷雾干燥法、反应结晶法等[25]。蒸发结晶法是从含有两种共结晶组分的过饱和溶液中进行溶剂蒸发,常用于制备晶态物质。冷却结晶法是样品在溶剂中溶解后,降温使溶液过饱和,然后使晶体析出的方法。喷雾干燥法将两种共结晶组分的不饱和溶液分散到喷嘴中,快速喷出溶剂后转化为固体粉末,在工业上常用于制备非晶态固体分散体,具有连续、快速和可控的特点,也可应用于共晶药物中共无定型药物合成[26]。WONG等[27]采用喷雾干燥法制备获得含有法匹拉韦与茶碱的无载体可吸入式共晶物粉末,改善了原药物的气溶胶性能和物理化学性质。该方法通过控制工艺,获得小粒度共晶粉末,方便呼吸给药,提高药物口服生物利用度和疗效。反应结晶法(或悬浮液法)是将过量的两种或两种以上组分加入单一/混合溶剂中直接反应形成难溶共晶。在同一溶剂中,若不同组分溶解度接近,一般按照1:1比例加入;溶解度差别较大时,可适当调整投料比;溶解度差别过大时,一般先将相对溶解度大的组分制成过饱和溶液,再加入另一组分。然而,由于共结晶过程中热力学和动力学作用比较复杂,且共晶纯度受多种因素影响,包括过饱和度、溶剂极性和CCF相对溶解度等,在应用于药物-药物共晶制备过程必须严格控制各种条件和参数。
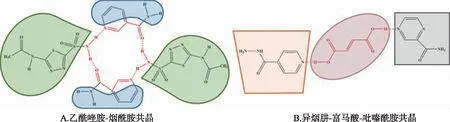
图2 基于不同类型的“超分子合成子”设计药物-药物共晶

表1 药物-药物共晶制备方法比较
2.3其他方法 超临界流体法:以超临界流体(一般为CO2)为介质,利用其溶剂动力和分子迁移增强作用来促进共晶形成的方法。RIBAS等[28-29]采用该法合成制备了姜黄素与烟酰胺、乙酰半胱氨酸两种共晶,提高了姜黄素的溶解率与抗炎活性。
种晶法:LIU等[30]研究发现,采用悬浮液易于制备获得索拉非尼与氟尿嘧啶共晶体,但其类似物瑞戈菲尼与氟尿嘧啶共晶体目前并不能通过干磨、液体助磨、溶剂浆化等方法制备。但当以索拉非尼-氟尿嘧啶共晶为籽晶,采用种晶法可制备得瑞戈菲尼与氟尿嘧啶共晶体,说明此类异种种晶策略有望促进新的药物-药物共晶的设计与发现。
热熔挤出法:采用机械化学技术,将固体或者半固体反应物放入热熔挤出机中,通过并排旋转的两根螺杆相互啮合研磨,使得反应物在受限的空间中被加热、剪切、压缩来促进共晶形成的方法。FERNANDES等[31]采用热熔挤出技术成功制备卡维地洛与烟酰胺共晶,有效改善了卡维地洛的溶解度与流动特性。目前热熔挤出法正逐渐应用于更多药物-药物共晶制备过程,并适于进行工艺放大和工业生产。
3 药物-药物共晶的应用优势
由于药物-药物共晶可以在不改变药物化学结构的基础上,克服与传统联合用药相关的问题,作为潜在的固体形式提供双重甚至多重药物治疗效果。同时可以有效调节相关API的理化性质,如改善药物溶解性、增加稳定性、改善生物利用度、预防多药耐药性和降低毒副作用等,且有助于增加现有产品的生命周期管理[32]。
3.1提高药物溶解性 药物溶解度能影响其生物利用度及通过胃肠道的渗透性[33],相关API的溶解度是影响药物吸收过程的基本因素之一。根据生物药剂学分类系统(biopharmaceutics classification system,BCS),有40%以上的药物是不溶或低水溶性的,药物-药物共晶技术是常用改善API溶解度的技术。
卡马西平是广泛应用的抗癫痫药物,属BCS Ⅱ类药物,水溶性差且体内生物利用度低。临床通常需选择较大剂量才能达到有效治疗效果。HUANG等[34]制备了卡马西平-大黄素和卡马西平-丹皮酚两种新型药物-药物共晶体,并考察了卡马西平原药及其共晶在水与pH值1.2和6.8的缓冲溶液中溶解度,结果表明卡马西平-丹皮酚在水中溶解度最大,并结合相溶解度图和理论计算解释了共晶调解卡马西平原药溶解度的机制。
相较其他传统β受体阻断剂如阿替洛尔、美托洛尔等,卡维地洛在降低患者血压等方面表现出更多优势,但低水溶性是其配方开发的主要障碍。EESAM等[35]设计卡维地洛与氢氯噻嗪形成共晶,在0.1 mol·L-1盐酸中共晶溶解度较卡维地洛提高7.3倍。
3.2改善药物稳定性 药物稳定性是评价药物质量的重要指标之一,API由活性物质转化为非活性代谢物会降低药物的治疗活性。奥沙利铂是第3代铂类抗癌药物,在水中不稳定,可诱导正常细胞产生毒性。YIN等[36]选择黄芩素和柚皮素为CCF,与奥沙利铂分别合成两种新型共晶。与奥沙利铂相比,共晶的溶出速率明显降低,具有更显著的抑制癌细胞作用。左氧氟沙星无水形式在室温条件下会迅速转化为半水合物和水合物形式,SHINOZAKI等[37]通过研磨与加热法制得左氧氟沙星与异乙酰氨基酚共晶体,降低了其吸湿性,提高了光稳定性。GOWDA等[38]选用悬浮液法得到依帕司他和二甲双胍共晶体,改善了依帕司他的溶解度和光稳定性,同时降低了二甲双胍的吸湿性,为二者联合应用提供了一种新方法。
3.3改善药物可压性 改善共晶物的机械性能可以改善药物的可压性,便于原料制备为合适的固体剂型。对于可压性较差的物质,还可以减少所需添加的辅料和(或)辅料的数量,从而减小片剂的总体尺寸,使片剂更容易吞咽,提高患者依从性[39]。盐酸二甲双胍具有吸湿性,且可能由于其层间的相互作用能各向同性和刚性,固态形式表现出较差的压片性能。水杨酸钠是一种非类固醇消炎药,压片性能明显好于二甲双胍[40-41]。BHATT等[39]在不采用任何片剂粘结剂的情况下,合成水杨酸钠与二甲双胍共晶,且在不同的压力下,该共晶表现出优越的压缩性、压实性和压片性。NASIR等[42]通过加液研磨法制得萘普生与烟酰胺共晶,改善了其力学性能。MODANI等[43]制备了非布索坦与吡罗昔康共晶,同时也评估了该共晶的可压缩性,结果表明其可塑性和直接压缩的适用性大大提高。
3.4提高药物生物利用度 先导物的低生物利用度是目前新药开发中瓶颈问题之一,药物-药物共晶技术为合成具有高水溶性与生物利用度的药物提供了新思路。由于异烟肼具有严重的肝毒性,LIU等[44]选择具有护肝作用的槲皮素与其合成共晶,通过体内外研究发现,共晶中槲皮素与其原药相比,溶出速率提高51.67倍,同时共晶在大鼠体内的口服生物利用度提高了28.91倍,基本清除了异烟肼所致肝毒性。贝沙罗汀是美国食品药品管理局(FDA)批准的抗肿瘤药物,具有分子量较小、脂溶性较高等优点,但低生物利用度限制了其临床治疗应用。REN等[45]制备贝沙罗汀和川芎嗪共晶体,并通过大鼠体内研究证明了所得共晶改善了贝沙罗汀的药动学性质,生物利用度明显提高。
3.5促进药物的协同作用 药物-药物共晶在增强药物理化性质、优化药动学性质的基础上,最显著的优势是可以发挥不同药物品种的协同作用。双靶点共晶药物沙库巴曲-缬沙坦(诺欣妥)就是药物发挥协同作用的典型代表。沙库巴曲是脑啡肽酶抑制剂的前体药,能够抑制脑啡肽酶,发挥舒张血管、促尿钠排泄等作用,但不能单独成药,必须同时阻滞肾素-血管紧张素-醛固酮系统(renin angiotensin aldosterone system,RAAS)。缬沙坦通过抑制血管紧张素Ⅱ 1型受体(anti-angiotensin II type 1 receptor,AT1R)对RAAS的作用,起到舒张血管、改善水钠潴留和减轻心脏负荷等作用。通过单晶X射线衍射分析发现,此共晶药物一个不对称单元由6个沙库巴曲分子、6个缬沙坦分子、18个钠离子和15个水分子组成。与传统治疗心力衰竭药物相比,共晶体中分子离子间存在复杂的氢键作用力和离子相互作用,有效改善了溶出速率,在体内同步发挥多重药效作用。该共晶药物在抑制脑啡肽酶的同时阻断AT1R,通过多重降压机制协同产生扩张血管、降低血压、排钠利尿、降低心脏负担等作用,在临床应用中疗效确切且安全性良好[46]。LV等[47]制备了氟尿嘧啶与山奈酚共晶,改善了原有单一的氟尿嘧啶口服吸收不完全且不均匀、代谢速度快和山奈酚溶解性差、口服生物利用度低等问题,提高了山奈酚稳定性,并优化了其溶出行为,在抗肿瘤中发挥协同作用。
4 结束语
近年来,药物-药物共晶技术在调节药物性质方面取得了巨大研究进展,例如溶解度、溶出速率、生物利用度、稳定性和可压性等。与其他晶体工程技术相比,共晶技术可使一些因出现新替代品而面临淘汰的“老药”重新获得利用。同时,作为一种有效的口服药物组合配方开发的新形式,有望克服原有药物的缺点。尽管药物-药物共晶体在制药领域引起了研发人员越来越多的兴趣与关注,但其开发过程仍面临着许多挑战。如共晶的设计和合成、筛选机制预测等都存在一定局限,需要进一步探索研究。但可以肯定的是,随着对该领域更加深入的研究,将会更多有效的药物-药物共晶药物在临床上发挥更大的作用。
猜你喜欢
选煤技术(2022年2期)2022-06-06
石材(2022年1期)2022-05-23
中学生数理化·中考版(2022年12期)2022-02-16
军事文摘(2020年18期)2020-10-27
石材(2020年2期)2020-03-16
模具制造(2019年3期)2019-06-06
含能材料(2017年1期)2017-03-04
含能材料(2017年7期)2017-03-04
中学化学(2016年10期)2017-01-07
当代化工研究(2016年6期)2016-03-20
