
镁掺杂煤基活性炭对亚磷酸的吸附性能研究
2023-06-25 01:39王佩文杨春明张壮壮杨晓霞甄延忠
延安大学学报(自然科学版) 2023年2期
高 飞,王佩文,杨春明,张壮壮,杨晓霞,甄延忠*,付 峰
(1.延安大学 石油工程与环境工程学院;2.延能-延大综合能源技术研究院;3.油气资源高效开发与生态环境保护陕西高校工程研究中心,陕西,延安 716000)
草甘膦是全世界使用最广泛的除草剂,作为草甘膦生产大国,中国每年的草甘膦产量约占全球的80%以上[1]。然而,生产草甘膦会伴随大量高亚磷酸含量(约5%)的草甘膦母液的产生,若这些高亚磷酸含量的母液处理不彻底就肆意排放,不仅会使得磷酸盐在水体富集造成水体富营养化和水体恶化,同时高值亚磷酸的流失会造成巨大的经济损失[2-3]。因此,回收草甘膦母液中的亚磷酸不仅能减少环境污染而产生良好的生态效益,还可以实现高值磷化物的回收而创造巨大的经济效益。
目前,水体中磷化物的去除与回收的主要方法有化学沉淀、生物降解、高级氧化、膜分离及吸附法[4]。其中,吸附法由于其经济、无二次污染、操作工艺简单、高效率等优点,一直被视为最有前景的方法[5]。吸附法通过吸附剂对吸附质产生较强黏附力和其他相互作用力,以实现从溶液中去除吸附质的目的。常见的去除工业废水中磷化物的吸附材料有:沸石、树脂、富含多金属的污泥、铁的氧化物等[6]。在选择吸附剂时,除了考虑其对磷化物的选择性、灵敏性和亲和性外,还应该考虑原料本身的优势以及制备的成本和对环境的友好性[7-8]。中国具有“富煤、贫油、少气”的自然能源禀赋,以储量丰富的煤炭为原料制备高附加值的功能炭材料,是实现煤炭资源高值化利用的必要途径[9-10]。目前,诸如煤基石墨碳、煤基活性炭等煤基碳材料在新兴产业的应用愈加广泛。因此,探索以煤为原料的碳材料用以吸附磷化工母液中的高值磷化物具有重要意义。
煤基活性炭材料由于具有良好的稳定性和发达的孔隙结构常作为吸附剂被广泛用于吸附领域。例如,谢克昌院士课题组利用高硫煤进行活化促进孔的发育以及金属掺杂的方式制备硫自掺杂多孔炭脱汞吸附剂并取得了良好的效果[11]。然而,煤基碳材料对草甘膦母液中亚磷酸等高值磷化物选择性分离的研究鲜有报道。基于此,以延安能源集团禾草沟煤为原料,通过KOH活化和浸渍法获得了镁离子负载的煤基活性炭材料(Mg-AC)。通过模拟亚磷酸溶液的吸附试验对比了改性前后的吸附性能,分析了pH 对亚磷酸吸附的影响;通过典型的动力学和等温线模型对实验数据进行拟合。该研究不仅为回收草甘膦母液中亚磷酸提供了工业化应用新思路,也丰富了煤基碳材料家族并为其提供了新的应用途径。
1 实验部分
1.1 主要材料与试剂
禾草沟煤(HCGM)样品取自陕西省子长市永兴煤矿;氢氧化钾(KOH),国药化学试剂有限公司;硝酸镁(Mg(NO3)2·6H2O)、盐酸(HCl,36%~38%),四川西陇化工有限公司;亚磷酸(H3PO3),国药集团化学试剂有限公司;过硫酸钾(K2S2O8)、钼酸铵((NH4)6Mo7O24·4H2O),上海阿拉丁生化科技有限公司。以上药品均为分析纯,实验室所用水均为超纯水,乙醇为无水乙醇。
1.2 主要仪器
XRD-7000 型全自动X 射线粉末衍射仪和UV-2500型紫外-可见分光光度计(日本岛津公司);JSM-7401F 扫描电子显微镜(SEM)和JEOL-F200 型透射电子显微镜(日本电子公司);ASAP 2020 型比表面积和孔隙度分析仪(美国麦克仪器公司);Nicolet-5700傅里叶变换红外光谱仪(日本电子公司);Nano ZS90 型纳米粒度电位仪(思百吉仪器公司);DGLS-35B高温高压灭菌锅(上海力辰仪器公司)。
1.3 材料制备
称取6 g 粒径为0.106~0.075 mm 的HCGM 浸渍于20 mL KOH 水溶液中,在25 ℃下磁力搅拌5 h后置于70 ℃烘箱中干燥12 h;称取适量干燥后的样品置于惰性气氛(N2)管式炉中,以10 ℃/min的升温速率加热至800 ℃,热解60 min 后自然冷却至室温;用1 mol/L HCl 溶液反复洗涤抽滤后用超纯水清洗至中性,最后将样品置于70 ℃烘箱中干燥24 h并过140目筛,即制得煤基活性炭(AC)。
配置250 mL 8 g/L 的Mg(NO3)2溶液备用,取4 g AC 加入到80 mL Mg(NO3)2溶液中,在25 ℃下以6 000 r/min 的转速磁力搅拌10 h 后抽滤,将滤出物在105 ℃烘箱中干燥8 h;最后取3.0 g干燥好的样品置于惰性气氛(N2)管式炉中,于550 ℃下热解30 min 后自然冷却,研磨即可得到镁掺杂煤基活性炭(Mg-AC)。
1.4 吸附实验
1.4.1 亚磷酸标准曲线
分别配置50 和100 mg/L的亚磷酸溶液各500 mL备用,采用GB 11893-89 钼酸铵分光光度法测定亚磷酸浓度,绘制标准曲线如图1所示,标准曲线方程为y=0.012 44x-0.139 02,R2=0.998。

图1 亚磷酸标准曲线
1.4.2 静态吸附实验
取0.10 g AC加到50.0 mL的亚磷酸溶液(100 mg/L)中,置于恒温振荡器中震荡2 h(25℃,150 r/min),分别在5、10、15、20、30、40、60、80、100、120 min 时取上清液测定亚磷酸浓度,用式(1)计算AC 吸附剂的吸附容量,同理得出Mg-AC的吸附容量。
其中,qe为平衡时吸附剂对亚磷酸的吸附容量(mg/g);V为溶液体积(mL);c0和ce分别为吸附初和吸附平衡时的溶液浓度(mg/L);m为吸附剂用量(g),此处为0.1 g。
1.4.3 吸附动力学模型
为对Mg-AC 吸附亚磷酸过程进行动力学研究,分别采用准一级动力学、准二级动力学、Elovich及Weber-Morris 模型拟合实验结果,模型公式如下:
其中,qt为t时刻Mg-AC对亚磷酸的吸附容量(mg/g);t为吸附时间(min);k1为准一级动力学的速率常数(1/min);k2为准二级动力学的速率常数[g/(mg·min)];kp为颗粒内扩散常数[g/(mg·min)1/2];α、β为吸附参数(mg/(g·h)、g/mg),Ci为与边界层厚度相关的常数(mg/g)。
1.4.4 等温吸附实验
将0.10 g Mg-AC 加入到50.00 mL 不同初始浓度(20、40、60、80、100 和120 mg/L)的亚磷酸溶液并置于恒温振荡器中,在25 ℃条件下恒温震荡120 h以上,测定达到吸附平衡状态下的亚磷酸浓度。采用Langmuir、Freundlich 和Temkin 模型拟合吸附平衡等温线,模型公式如下:
其中,ce为亚磷酸溶液平衡时浓度(mg/L);qm为最大单分子层吸附容量(mg/g);KL为Langmuir常数(L/mg);KF为Freundlich常数(mg/g);KT为Temkin模型常数(L/g);1/n为Freundlich吸附强度参数。
1.5 样品表征
合成样品的物相组成可采用X 射线衍射仪进行测定,以Cu Kα(Ni 滤波片,λ=0.154 18)为靶源,管电压为40 kV,测试范围2θ为10°~80°,扫描速度为8°/min;采用傅里叶变换红外光谱仪分析HCGM 和Mg-AC 的表面官能团,测试波数范围为400~4 000 cm-1,分辨率为4 cm-1,扫描15 次;采用扫描电子显微镜和透射电子显微镜分析样品的粒度尺寸、孔隙等表观形貌特征,并测试了N、O、Mg 的元素分布图;采用比表面积和孔隙度分析仪测定改性前后样品的比表面积及孔径;采用X-射线光电子能谱仪检测吸附剂的光电子能谱,铝靶激发源采用Al Kα 射线(hv=1 486.68 eV),工作电压为14.6 kV,以C1s 为标准进行荷电校正;采用纳米粒度电位仪在25 ℃条件下测试样品表面电位。
2 结果与讨论
2.1 活性炭表征
2.1.1 表观形貌分析
图2 为煤基活性炭样品的SEM 图。由图2A 可知,原始禾草沟煤是由直径为5 μm 左右的不均匀的块状堆叠而成,且结构简单、表面光滑。经KOH活化后得到AC 表面粗糙且存在大量孔径不一的蜂窝状孔隙,如图2B 所示。从图2C 可以看出Mg-AC表面孔隙结构复杂、粗糙且不规则,部分孔内有颗粒填充物,表明Mg2+与AC 表面官能团发生化学反应并负载在其表面。图2D为Mg-AC的TEM 图,可知AC经Mg(NO3)2浸渍之后依旧是块状结构,说明其结构稳定,不易被破坏。图2E、2F 和2G 分别为Mg-AC的C、O和Mg元素的分布图,可以看出C、O、Mg都均匀分布在样品表面,表明通过Mg(NO3)2浸渍成功地将Mg掺杂到AC表面。
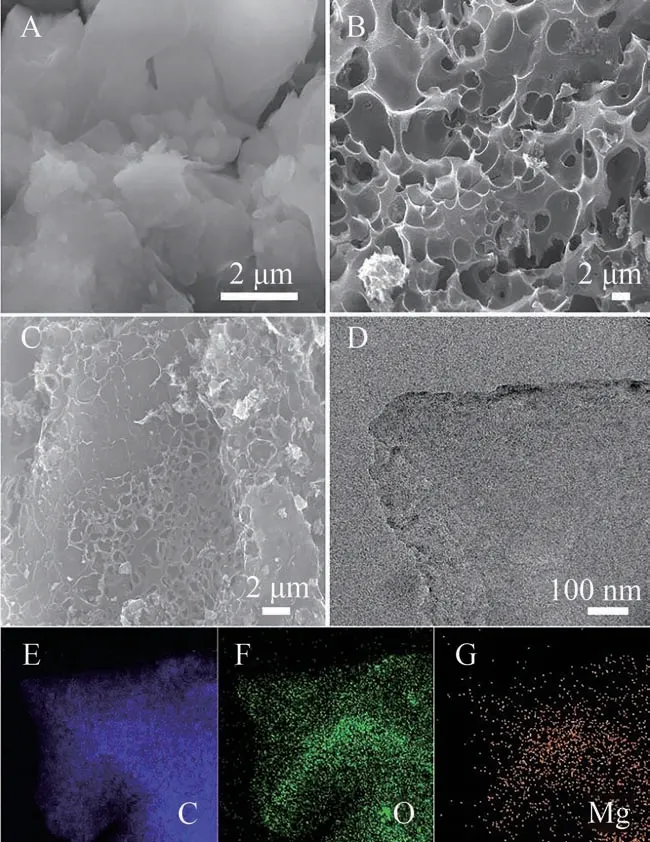
图2 样品(A-C)的SEM图;样品C的TEM图(D)和元素分布图(E-G)
2.1.2 物相及表面官能团分析
图3A 为样品的XRD 图谱,可知HCGM 中杂质SiO2和CaCO3经KOH活化之后被去除,得到的AC能观察到Al2O3的峰,这可能是由于比表面积和孔隙增大后,检测到Al2O3样品拖的峰。AC 和Mg-AC 均在23°和43.5°处存在明显的衍射峰,分别对应于石墨结构的(002)晶面和(100)晶面,表明AC和Mg-AC中存在石墨微晶,且二者均为石墨化程度较低的无定型碳[12]。相较于AC,Mg-AC的(100)晶面变化不明显,而(002)晶面衍射强度明显变弱,趋于平缓,说明浸渍掺杂镁并高温热解会对AC中规则的晶体结构造成一定缺陷,使得Mg-AC石墨化程度有所降低,碳材料无序度增大[13]。
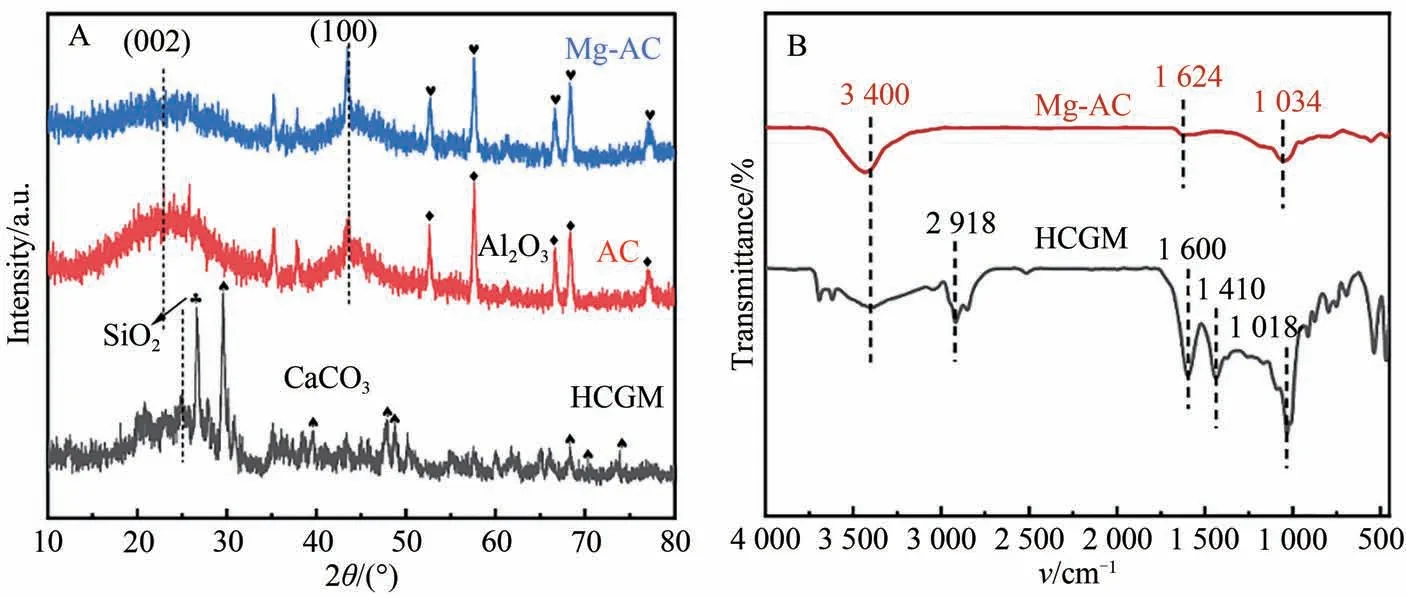
图3 (A)样品的XRD图谱;(B)样品FT-IR图谱
图3B 为样品的FT-IR 图谱,可知Mg-AC 在3 000~3 700 cm-1处有较宽的吸收峰,可归因于存在—OH或—CHO官能团,表明Mg-AC表面存在—OH官能团。1 624 cm-1处为芳香族的C=C或酮和酰胺中的C=O 官能团的伸缩振动所产生的特征峰,1 034 cm-1处的特征峰对应于C—O的伸缩振动[14-15]。Mg-AC 的红外光谱与HCMG 有明显差异,1 600 和1 018 cm-1处的特征峰向1 624和1 034 cm-1处偏移且峰强度减小,在1 410 cm-1处的特征峰几乎消失。这可能是由于负载的Mg2+与活性炭上的官能团(—OH、—C=O 和—COOH)发生络合作用,形成更复杂的—(COO)n—Mg,—(CO)n—Mg 和—On—Mg 等含镁官能团[16]。
2.1.3 比表面积和孔结构分析
图4A 为样品的N2吸附-脱附等温线,可知HCGM 的吸附等温线为类型Ⅲ[7],说明HCGM 为典型的无孔结构,比表面积不足4.0 m²/g。而经KOH 活化制备的AC 以及经Mg(NO3)2浸渍并热处理后的Mg-AC 样品的N2吸附-脱附等温线均为结合型的Ⅰ型和Ⅳ型[17]。当p/p0<0.1 时,等温线迅速上升,说明样品存在微孔,结合图4B 样品的孔径分布图,表明Mg-AC 表面存在以40 nm 为中心的大量介孔。当p/p0>0.1 时,每步的吸附量缓慢增加,同时当p/p0>0.45 时,AC 及Mg-AC 的吸附和脱附等温曲线形成H4 型回滞环,展示了一个典型的介孔特征[15],揭示AC 和Mg-AC 是微孔-介孔材料。由于比表面积和孔隙结构是影响吸附剂吸附能力的重要因素,结合表1 可知,与HCGN 相比,AC 的比表面积(S)由3.1 m2/g增加到820.8 m²/g,相应的平均孔容(V)也由0.012 cm3/g增加到0.502 cm3/g,而较大的比表面积和孔容有利于为吸附亚磷酸提供更多的活性位点[18]。Mg(NO3)2沉积在大中孔内会略微破坏AC 的孔隙结构,提高吸附剂表面粗糙度,使得Mg-AC 比AC 的比表面积稍微增大(由820.8 m²/g 增大到896.4 m²/g)。

表1 样品的孔结构参数
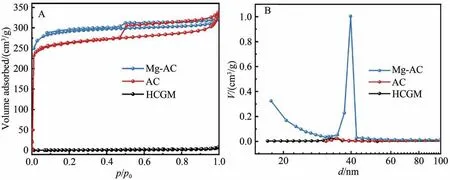
图4 (A)样品的N2吸附-脱附等温线;(B)样品的孔径分布图
2.1.4 光电子能谱分析
HCGM、AC 以及负载Mg 后的活性炭Mg-AC 的XPS全谱图如图5A所示,图5B~5D分别为HCGM、AC和Mg-AC 样品的C1s 分峰结果。由图5A 可知,三者均含有大量的C、O元素和微量的N、S元素,Mg-AC含有少量Mg元素,证实了Mg被成功掺入到AC表面。在C1s 精细谱分峰图中,C—C、C—O—C/C—OH 和O=C—O 官能团对应特征峰的结合能范围依次为284.4~284.8 eV、285.0~286.3 eV和288.4~289.9 eV[18-19]。结合禾草沟煤改性过程中活性炭的不同碳形态及其相对含量变化(表2)可知,在HCGM 经KOH 活化得到AC 过程中,C—C 的相对含量由61.76%降至56.88%,O=C—O 的含量由13.61%增至18.30%。说明KOH 在活化过程中消耗了C—C,致使其相对含量降低,并引入了含氧官能团,与HCGM相比,AC和Mg-AC 的C—O—C/C—OH 和O=C—O 的相对含量均增加。
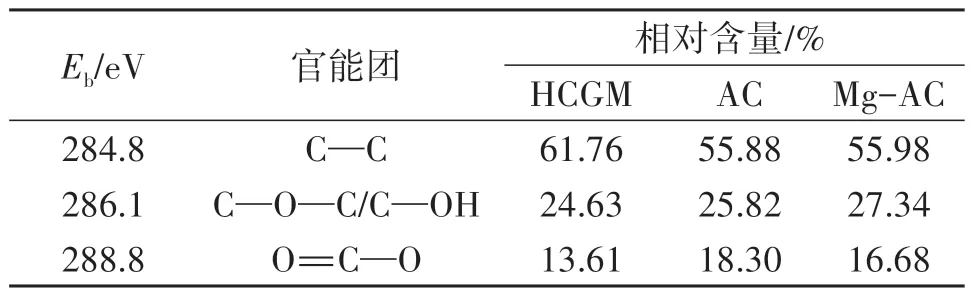
表2 样品的碳形态和相对含量
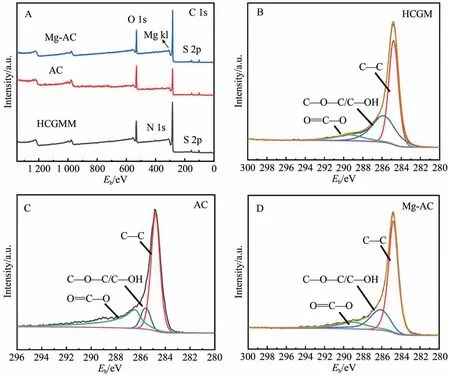
图5 (A)样品的XPS全谱图;(B~D)HCGM、AC、Mg-AC的C1s高分辨谱
2.2 吸附性能
2.2.1 样品吸附能力
图6A 为HCGM、AC 和Mg-AC 对亚磷酸的吸附容量随时间的变化。可知HCGM 对亚磷酸几乎无吸附能力,AC 对亚磷酸展示出一定的吸附能力,吸附平衡时吸附容量不足3.0 mg/g。相较于AC,Mg-AC 对亚磷酸的吸附容量数倍增加,达到吸附平衡时吸附容量约为15.0 mg/g,这是由于改性后镁成功负载到AC 表面及孔隙结构内,有利于促进亚磷酸根离子与含镁化合物产生分子间作用力,与Mg2+中的空d 轨道发生络合作用,对亚磷酸展现出良好的吸附能力。

图6 (A)样品吸附亚磷酸吸附容量的变化;(B)样品的Zeta电位图;(C)pH对Mg-AC吸附亚磷酸的影响
2.2.2 pH的影响
pH 通过改变吸附剂表面电负性和电荷数量影响吸附剂和吸附质间的静电作用进而影响吸附过程,如图6B 所示,当初始pH=2 时,Mg-AC 由于质子化作用表面带正电,且电势约为25 mV,可与亚磷酸根阴离子之间产生静电引力。采用0.1 mol/L的HCl和NaOH 溶液调节溶液pH 值(2~12)以探究pH 对Mg-AC吸附容量的影响,结果如图6C所示,当pH值由2 增加至3 时,Mg-AC 对亚磷酸的吸附容量基本成倍增大;当pH 范围为3~7 时,吸附容量稳定在13.0 mg/g 左右;当pH 为7~12 时,吸附容量逐渐减小;综合而言,当pH 范围为3~10 时,Mg-AC 对亚磷酸的吸附容量能达到10.0 mg/g 以上,该吸附剂具有较宽的pH应用范围。
2.2.3 吸附动力学
利用常见的3 种动力学方程(式(2)~(4))拟合动力学实验结果,利用颗粒内扩散模型(式(5))将吸附过程分为3 个阶段,结果如图7 所示。由图7A及表3 可知,准一级、准二级动力学和Elovich 模型均能较好地拟合动力学吸附过程,相对地,准二级动力学模型拟合的相关系数R2=0.991,能更好地拟合吸附过程,且理论平衡吸附容量为16.3 mg/g,吸附速率与亚磷酸浓度成正比,k2=0.012 3 g/(mg·min),展示出良好的吸附速率,说明吸附过程以化学吸附为主。

表3 Mg-AC吸附亚磷酸动力学拟合参数
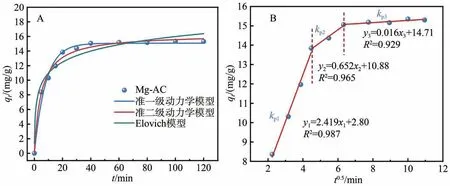
图7 (A)Mg-AC的吸附动力学拟合;(B)颗粒内扩散模型
由图7B 可知,吸附过程可分为3 个阶段,第一阶段为瞬时吸附阶段,发生在Mg-AC 外表面,吸附速率快,基本在20 min 内完成,归因于起初Mg-AC表面具有大量吸附位点以及存在较大的亚磷酸根离子浓度差,传质推动力较大,此时吸附容量为13.9 mg/g,约达到饱和吸附容量的90%。第二阶段描述吸附分子在吸附剂中的扩散过程,此过程与孔体积和尺寸有关,kp2<kp1,相比于第一阶段,吸附速率减缓,吸附逐渐趋于平衡状态。第三阶段在40 min 之后,为吸附平衡阶段,此时吸附质浓度较低,吸附容量基本不变,约为15.3 mg/g。颗粒内扩散模型图分为3 段,说明Mg-AC 吸附亚磷酸的过程复杂,且速率常数的变化说明颗粒内扩散和外表面扩散共同控制着吸附速率。鉴于颗粒内扩散模型拟合直线不经过原点,说明颗粒内扩散并不是唯一的控制因素,可能还存在静电作用等,与PH影响实验结论一致。
2.2.4 吸附等温线
利用常见等温吸附模型(式(6)~(8))对亚磷酸在Mg-AC的吸附平衡过程进行数据拟合,结果如图8 所示。可知随着亚磷酸初始浓度的增大,Mg-AC 的吸附容量也随之增大。因为当初始浓度逐渐增大时,亚磷酸更容易进入Mg-AC 孔隙中,增大了亚磷酸和Mg-AC表面活性位点的接触几率,此时吸附速率较快;而当初始浓度较高时,吸附亚磷酸的活性位点已趋于饱和状态,使得未吸附的亚磷酸对吸附位点的竞争加剧,进而吸附容量的增速变缓。结合等温吸附模型拟合参数(表4)可知,Langmuir、Freundlich 和Temkin 模型都能很好地拟合Mg-AC吸附亚磷酸的过程,R2分别为0.970、0.900 和0.968。相对地,Langmuir模型的相关系数R2=0.970,最大吸附容量为14.2 mg/g,与实验值(15.3 mg/g)更为接近,表明Mg-Ac 对亚磷酸的吸附更符合Langmuir 模型,可主要用均匀表面和单分子层吸附去描述吸附过程。

表4 吸附等温线拟合参数
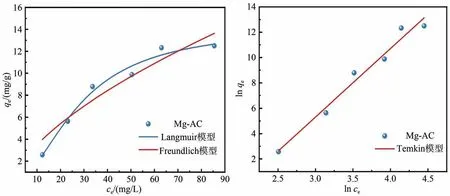
图8 Mg-AC的吸附等温线
3 结论
以禾草沟煤为原料,通过KOH活化法改性制备AC,通过Mg(NO3)2浸渍并热解得到镁掺杂的活性炭Mg-AC。研究表明,经KOH 活化的AC 比表面积、总孔体积均大幅增加,经Mg2+浸渍并热解制备的Mg-AC 较AC 对亚磷酸的吸附容量数倍增加,pH 范围为3~10时,吸附容量能达到10.0 mg/g以上。吸附动力学和等温吸附模型拟合结果表明,Mg-AC 对亚磷酸的吸附过程更符合准二级动力学模型(R2=0.991)和Langmuir模型(R2=0.970),说明吸附过程以化学吸附和单分子层吸附为主导。吸附是通过孔扩散、络合作用和静电引力的协同作用吸附亚磷酸,且饱和吸附容量为15.3 mg/g。由于合成Mg-AC 原材料获取便捷,制备工艺简单,且对亚磷酸具有优越的吸附性能,因而极具实际应用潜力。
猜你喜欢
化工管理(2022年13期)2022-12-02
中国特种设备安全(2021年4期)2021-10-13
能源工程(2021年1期)2021-04-13
无机盐工业(2020年6期)2020-06-12
山东化工(2020年4期)2020-03-30
中国塑料(2015年8期)2015-10-14
中学政史地·教学指导版(2014年10期)2015-02-02
食品工业科技(2014年15期)2014-03-11
河南科技(2014年7期)2014-02-27
发明与创新(2013年13期)2013-03-11
