
TGF-β通过调控线粒体功能影响肺癌细胞的侵袭和迁移
2023-06-20 08:28郑宗耀陈智鹏曾观娣
暨南大学学报(自然科学与医学版) 2023年2期
郑宗耀,陈智鹏,曾观娣
(暨南大学 生命科学技术学院 肿瘤分子生物学教育部重点实验室,广东 广州 510632)
肺癌仍是我国乃至世界发病率和死亡率最高的癌症[1-2]。肺癌可分为非小细胞肺癌和小细胞肺癌,其中非小细胞肺癌占80%~85%。目前已知肺癌发生主要与致癌基因(如EGFR及KRAS突变、ALK重排)的改变有关[3]。因此,驱动基因的鉴定与药物开发是肺癌研究领域的重点[3]。尽管靶向疗法在一些患者身上已取得了良好效果,但是仍有相当一部分患者对靶向药物响应不佳或者产生耐药。因此,肺癌的发病及耐药产生的确切机理仍需阐明。
肺癌异质性是当前治疗未响应及耐药产生的潜在原因[4-5]。癌症干细胞(cancer stem cells,CSCs),除了有活跃的端粒酶活性,可能导致基因不稳定,发生癌变;还有无限制且高效的增殖能力,其所在的组织、器官等部位自我更新率越快,肿瘤的发生率也越高;并且与肿瘤起始、恶性发展、耐药有关[6-7],是引起实体瘤异质性的重要原因。近年来研究发现上皮间质转变(epithelial-mesenchymal transition,EMT)可促进癌细胞干性和肿瘤起始能力,因此被认为是CSCs的来源之一[8-9]。另外,EMT赋予癌细胞迁移、侵袭、抗凋亡和耐药的能力,增加了治疗难度[8]。
转化生长因子-β(transforming growth factor-β,TGF-β)是EMT主要调控因子,主要通过经典Smad依赖通路和非经典Smad通路,激活EMT相关的转录因子和miRNAs,改变表观遗传特征,诱导上皮细胞EMT,获得干性和耐药能力[10-14]。另外,TGF-β信号受到Smad7蛋白、mRNA可变剪切和翻译后修饰等调控[11]。而且TGF-β在肿瘤中与相关的免疫微环境息息相关,影响分化淋巴前体的命运和肿瘤内多个白细胞亚群的活性[15]。由此可见,TGF-β的分化信号涉及多个通路的相互协调作用。因此,在此过程中仍存在未知的调控机制。
线粒体是细胞物质能量代谢和信号中心,参与调控包括代谢、增殖、分化等多个细胞进程[16]。另外,线粒体形态和质量控制与其不同功能和活性的发挥有密切联系[17]。在癌症中,生长因子和线粒体基因突变等因素可引起线粒体代谢功能的重编程,而线粒体所反馈的信号可调控相应的信号通路,影响染色质结构、转录和翻译,促进癌细胞恶性发展,并且线粒体控制生物体能量代谢,其功能受损容易导致高乳酸环境产生,促进癌症进展[16,18]。目前线粒体与癌细胞EMT的关系尚不清楚,本研究基于TGF-β诱导的肺癌A549细胞EMT模型,探索线粒体在细胞接受分化信号时的变化和线粒体对细胞发生EMT的影响,并阐明其作用机制。
1 材料与方法
1.1 试剂与仪器
A549细胞购自美国典型培养物保藏中心ATCC。HEK293T细胞购自中国科学院上海细胞库。包装质粒psPAX2、pMD2.G、pLKO-tet-puro购自Addgene。pLVX-TetOne-Puro质粒来源于Clontech。pGL3、Renilla荧光素酶表达质粒和pRL-TK海参荧光素酶表达质粒来源于实验室自保存。购买的其他试剂包括:兔抗人Smad2和E-cadherin单克隆抗体(CST);鼠抗人vimentin(Santa)和β-actin(Sigma)单克隆抗体;鼠和兔HRP二抗(sigma);Dual-Luciferase® Reporter Assay System(Promega);Trizol(Invitrogen);TGF-β抑制剂SB431542(MCE);PI3K 抑制剂LY294002(MCE);AKT抑制剂MK-2206(MCE);RT试剂盒(爱柏梦);SYBR green核酸染料(Monad);Lipofectamine 3 000(Invitrogene);胎牛血清(Gibco);RPMI-1640培养液和DMEM培养液(Gibco);PBS磷酸盐缓冲液(Servicebio);蛋白酶、磷酸酶抑制剂(Biotech Roche);RIPA蛋白裂解液(碧云天);线粒体膜电位检测试剂盒(JC-1)(碧云天);四环素(DOX,Solarbio);BCA试剂盒(Thermo Fisher Scientific);Western blot PVDF膜(Millipore);青链霉素抗生素(Gibco);胰酶(Gibco);嘌呤霉素(Sigma);ECL超敏化学发光试剂(Millipore)。
主要仪器:凝胶成像仪(FUSION FX);荧光定量PCR仪(Bio-Rad);流式细胞分析仪(BD)。
1.2 研究方法
1.2.1 细胞培养
实验所用细胞系均用新配制的RPMI-1640完全培养液(含10%胎牛血清、1%青链霉素抗生素)重悬细胞,经吹打混匀后接种于培养皿,置于37 ℃、5% CO2及饱和湿度培养箱内培养,每天更换培养液,倒置显微镜下观察细胞形态。待贴壁细胞完全生长汇合后,用0.25%胰酶(含0.02% EDTA)消化传代,待细胞扩增至一定数量后,收获细胞进行相关实验。HEK293细胞用新配制的DMEM完全培养液(含10%胎牛血清、1%青链霉素抗生素)培养。
1.2.2 载体构建
PGC-1α启动子表达质粒和DOX诱导表达的Smad2或PGC-1α敲低质粒按照基因表达载体标准构建方法构建。其中所用到的shRNA序列信息来源于Sigma Mission shRNA文库shPPARGC1A (TRCN0000364085和TRCN0000364084),shSMAD2(TRCN0000040035和TRCN0000040036),构建pLVX-tet-PPARGC1A-3XFLAG。所用引物序列为前引物:5’-TCCTACCCTCGTAAAGAATTCATGGA-TGAGACCTCCCCAAGG-3’;后引物:5’-GTCTTTG-TAGTCAGGCGCGCCCCTGCGCAAGCTTCTCTGAG-3’。
PGC-1α报告基因质粒构建,根据软件预测,人PGC-1α转录本1的转录起始位点前2 000 bp有2个潜在的SEB结合位点,因此通过PCR扩增其转录起始位点前2 000 bp序列并构建到pGL3载体。所用引物序列为前引物:5’-TTTCTCTATCGATAGGTACCATGTGTTAGGGCAA-ATAACCATGT-3’;后引物:5’-AGTACCGGAAT-GCCAAGCTTCTGAATGACGCCAGTCAAGC-3’。
1.2.3 慢病毒感染
将两种包装质粒(psPAX2和pMD2.G)和pLKO-tet-puro对照或相同用于表达PGC-1α或shSmad2、shPGC-1α分别一起转染HEK293细胞。24 h后,细胞再换新培养基培养24 h。收集上述含有慢病毒的培养液上清,0.45 μm滤膜过滤,病毒上清和A549细胞悬混液以体积比1∶1混合感染,期间添加polybrene(8 μg/mL)增加慢病毒感染效率。感染后的细胞经嘌呤霉素(0.5 μg/mL)筛选7 d,随后验证构建好的细胞稳转株中目的蛋白表达情况。
1.2.4 JC-1染色检测细胞膜电位
收集经药物处理后的细胞计数后,取(1~6)×105个细胞,重悬于0.5 mL细胞培养液中,加入0.5 mL JC-1染色工作液,颠倒数次混匀。细胞培养箱中37 ℃孵育20 min。孵育结束后,1 200 r/min 4 ℃离心3~4 min,沉淀细胞。弃上清,用预冷的JC-1染色缓冲液(1×)洗涤2次:加入1 mL JC-1染色缓冲液(1×)重悬细胞,1 200 r/min 4 ℃离心3~4 min,沉淀细胞,弃上清。再加入1 mL JC-1染色缓冲液(1×)重悬细胞,1 200 r/min 4 ℃离心3~4 min,沉淀细胞,弃上清。再用适量JC-1染色缓冲液(1×)重悬后,进行流式细胞术分析。
1.2.5 DCFH-DA染色检测细胞ROS水平
按照VDCFH-DA∶V无血清培养液=1∶1 000的比例稀释DCFH-DA,使终浓度为10 μmoL/L。细胞收集后悬浮于稀释好的DCFH-DA中,细胞密度为(1~20)×107/mL,37 ℃细胞培养箱内孵育20 min。每隔3~5 min颠倒混匀一下,使探针和细胞充分接触。用无血清细胞培养液洗涤细胞3次,以充分去除未进入细胞内的DCFH-DA,随后进行流式细胞术分析。
1.2.6 RT-PCR检测mRNA表达情况
使用Trizol试剂提取总RNA,并逆转录为cDNA,于-80 ℃保存。后续进行荧光实时PCR分析以测量指定基因的mRNA水平,qPCR的反应条件为①激活:50 ℃,持续2 min;②预浸泡:95 ℃ 10 min;③变性:95 ℃ 15 s;退火:60 ℃ 1 min;④熔解曲线:95 ℃ 15 s,60 ℃ 15 s,95 ℃ 15 s。数据所示的是标准化为β-Actin的相对mRNA表达水平。基因特异性引物序列见表1。采用2-ΔΔCt计算目的基因的相对表达量。

表1 RT-qPCR引物序列Table 1 The sequences of the primers for RT-qPCR
1.2.7 Western blot检测蛋白表达情况
全细胞裂解物通过使用RIPA裂解缓冲液裂解,其中各加入100×蛋白酶和磷酸酶抑制剂混合物(Roche),蛋白质量浓度通过BCA试剂盒测定。易溶蛋白(30~40 μg)经SDS-聚丙烯酰胺凝胶电泳,而后转移至PVDF膜,快速封闭液室温封闭1 h,经TBST(含0.1%吐温-20,下同)洗涤1次后分别4 ℃孵育一抗过夜,第2天TBST洗涤5次,5 min/次,再室温孵育二抗(V二抗∶V抗稀释液=1∶5 000)1 h,最后用ECL超敏化学发光试剂显影,凝胶成像仪曝光成像并分析灰度值。
1.2.8 报告基因实验
将HEK293细胞提前铺于6孔板,待细胞完全贴好后转染基于pGL3构建的PGC1启动子表达质粒2 μg,同时也转染50 ng pRL-TK载体作为转染效率控制。24 h后将细胞分为对照组和实验组接种于12孔板,对照组和实验组分别进行DMSO和TGF-β处理,最后用双荧光发光试剂盒和酶标仪检测。
1.2.9 划痕损伤修复实验
将细胞接种6孔板培养至单层汇合状态,用200 μL吸头尖每孔划平齐的横线,PBS洗去未贴壁细胞,加入含0.5%胎牛血清的RPMI-1640培养液,对照组不给予DOX诱导,实验组进行1 μg/mL DOX诱导,于培养0 h和48 h镜下观察并拍照,ImageJ 8.0软件分析划痕愈合程度。
1.2.10 Transwell迁移实验
将Transwell小室置于24孔板,细胞消化后用含0.5% 胎牛血清的RPMI-1640培养液重悬,取2×104个细胞接种于Transwell小室的上室,体积为200 μL。在Transwell小室的下室中加含10%胎牛血清的RPMI-1640培养液500 μL,对照组不给予DOX诱导,实验组进行1 μg/mL DOX诱导,置于37 ℃、5% CO2、饱和湿度的培养箱中继续培养24 h,培养结束后取出小室并用4%中性甲醛固定15 min,0.1%结晶紫液染色5 min,用棉签小心擦除小室上室面细胞,PBS洗涤后在显微镜镜下观察并拍照。以穿膜的细胞数差异代表细胞运动能力的改变。每张膜选2个典型视野,每组重复3孔。
1.2.11 CCK-8实验
取提前准备好的生长状况良好的细胞,将其用胰酶消化,培养基重悬,制成单细胞悬液。细胞计数之后,将其平均分配到96孔白色透明板中。将细胞分为对照组及DOX组,DOX组中加入1 μg/mL DOX。每孔1 000个细胞、100 μL的培养基,于37 ℃、5% CO2条件下培养,每孔加入10 μL CCK-8试剂后孵育1 h,酶标仪读取各孔吸光度D(450 nm),此后每24 h按此法检测1次,每组设3复孔,取均值。实验设置天数为6 d。
1.2.12 Sphere实验
取提前准备好的生长状况良好的细胞,将其用胰酶消化,培养基重悬,制成单细胞悬液。细胞计数之后,将细胞按照实验需要进行分组,并且按照300个/孔接种到低贴附性的6孔板中,6孔板中需要提前加入配置好的sphere培养(DMEM/F12 培养基,20 μL/mL B27,20 ng/mL FGF和20 ng/mL EGF),放置在37 ℃ CO2培养箱中培养2周,期间每5 d添加相应的sphere培养基和DOX(1 μg/mL),每组实验设置3个复孔。待到球体长成后,轻轻摇匀培养皿将球体聚拢到中央,然后将其置于显微镜下拍照记录,其中取球体直径到达200 nm以上的细胞为计数标准。
1.3 统计学分析
用GraphPad prism6统计软件进行分析。数据代表3个独立实验并且是通过非配对t检验分析或者One-way ANOVA分析,误差条表示SEM。以P<0.05为差异有统计学意义。
2 结果
2.1 TGF-β在诱导A549细胞EMT过程中对线粒体表型的影响
基于所报道过的TGF-β诱导A549细胞EMT的模型[19],开展实验以评价TGF-β处理对A549细胞线粒体表型的影响。结果显示,TGF-β可显著促进线粒体膜电位增加(P<0.05,图1A)。另外,细胞ROS水平也显著增加(P<0.01,图1B),过量ROS反过来会对线粒体造成损伤。进一步通过RT-PCR检测线粒体基因的表达,同样发现TGF-β显著抑制这些基因的表达(P<0.01,图1C)。上述实验结果证明TGF-β处理引起A549细胞线粒体表型改变,对线粒体的功能具有抑制作用。
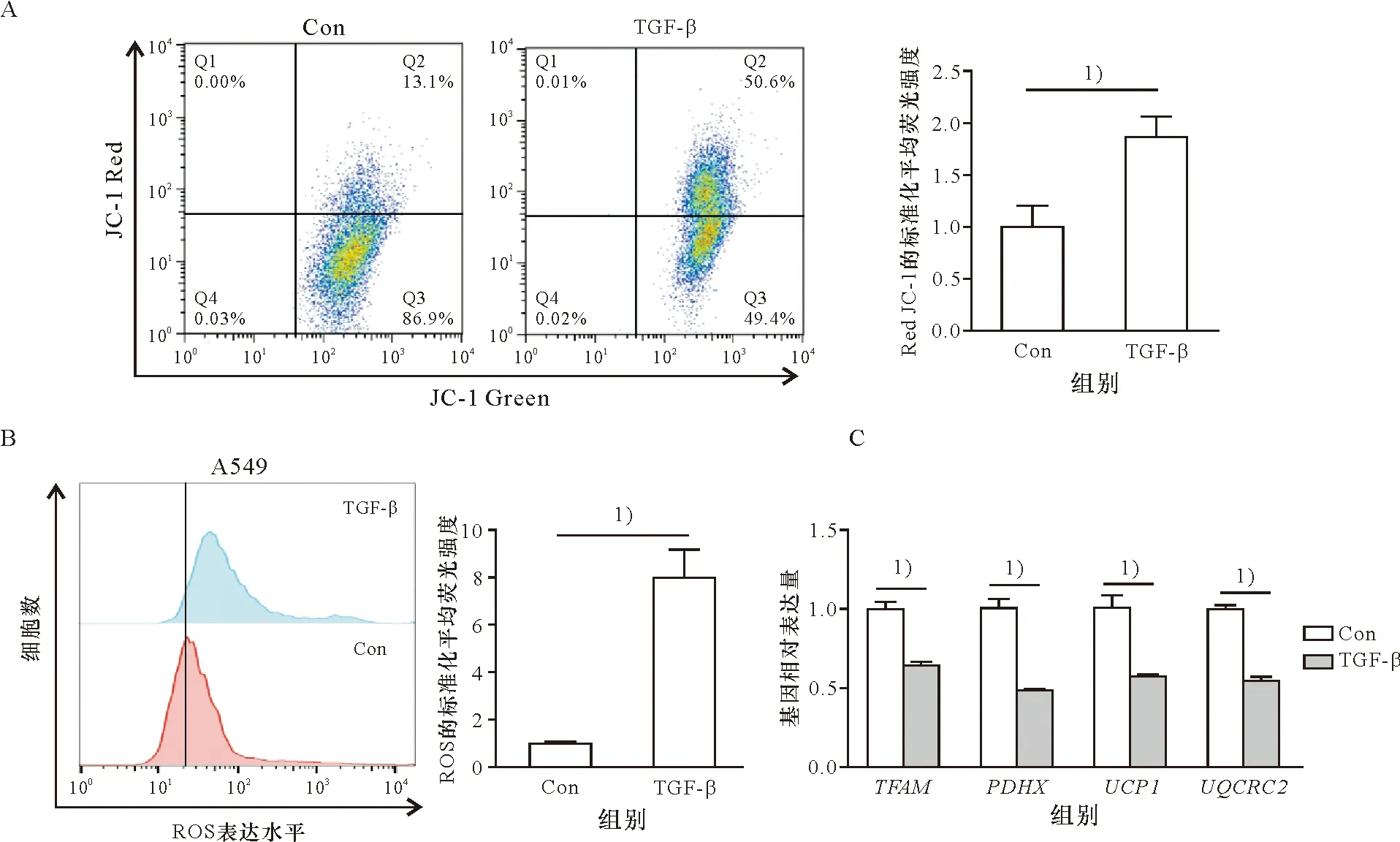
A:TGF-β刺激与否线粒体膜电位的比较;B:TGF-β刺激与否线粒体ROS水平的比较;C:TGF-β刺激与否线粒体基因表达的比较。1) P<0.05
2.2 TGF-β通过经典的Smad通路对线粒体表型的影响
为了确定TGF-β是通过Smad通路引起线粒体表型改变的,分别通过TGF-β受体抑制剂和Smad2敲低的方法对此现象进行验证。结果发现SB431542可显著抑制TGF-β引起的细胞ROS水平增加(P<0.01,图2A)。本研究构建了Smad2可诱导敲低的A549细胞株,在DOX诱导下可显著敲低A549细胞中的Smad2表达(P<0.001,图2B)。利用此细胞株,发现Smad2敲低后,可抑制TGF-β引起的细胞ROS水平增加(P<0.05,图2C)。进一步检测细胞线粒体基因的表达发现,Smad2敲低可恢复TGF-β引起的线粒体基因表达下调(P<0.01,图2D)。综上所述,TGF-β通过经典的Smad通路引起A549细胞线粒体表型改变。

A:各组线粒体ROS水平比较;B:蛋白质印迹评估构建的 A549-tet-shSmad2 细胞中的 Smad2 敲低效率;C:各组线粒体基因表达比较。1)P<0.05。SB:TGFBR抑制剂 SB431542
2.3 TGF-β通过经典的Smad通路对PGC-1α转录的影响
PGC-1α是调控线粒体功能及线粒体基因表达的一个重要基因,通过对mRNA水平的检测,发现TGF-β可显著抑制PGC-1α的表达(P<0.01,图3A)。利用本研究所构建的PGC-1α报告基因,发现TGF-β可显著抑制其转录活性(P<0.01,图3B)。进一步通过多个抑制剂的分析,发现PGC-1α的转录表达受经典Smad通路调控(图3C)。上述结果说明TGF-β通过经典的Smad通路抑制PGC-1α的转录表达而发挥作用。
2.4 PGC-1α恢复TGF-β引起的线粒体基因表达下调
为验证TGF-β是否可通过PGC-1α影响线粒体基因的表达,构建了PCG-1α可诱导表达的A549细胞株,结果显示DOX可显著促进PGC-1α表达增加(P<0.001,图4A)。在此细胞株中,当诱导PCG-1α表达时,可恢复TGF-β引起的线粒体基因表达下调(图4B)。以上实验结果说明TGF-β造成的细胞线粒体功能改变是通过影响下游效应分子PGC-1α的表达所致。
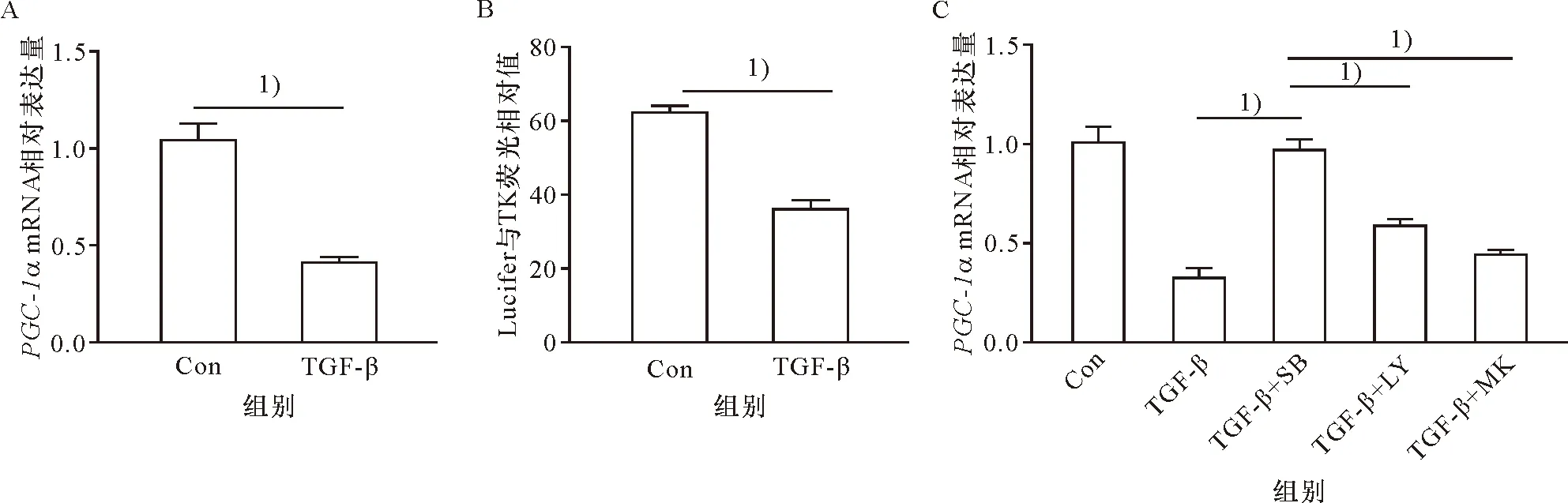
A:TGF-β处理与否的A549 PGC-1α mRNA水平比较;B:通过PGC-1α启动子报告基因测定评估PGC-1α在TGF-β处理中的转录活性;C:各组PGC-1α mRNA的表达比较。1) P<0.05.SB:TGFBR抑制剂SB431542,LY:PI3K 抑制剂 LY294002,MK:AKT 抑制剂MK-2206

A:通过RT-PCR评估构建的A549-tet- PGC-1α细胞中PGC-1α的表达;B:A549-tet-PGC-1α细胞中相应处理后各组间线粒体基因表达比较 1) P<0.05
2.5 PGC-1α敲低对A549细胞EMT的影响
为验证TGF-β抑制PGC-1α表达是促进A549细胞EMT的机制之一,构建了PCG-1α可诱导敲低的A549细胞株模拟TGF-β抑制PGC-1α表达的作用。首先,DOX可显著诱导PCG-1α表达下调(P<0.01,图5A)。通过免疫印迹实验,发现PCG-1α诱导敲低后引起细胞上皮标记物E-cadherin表达显著下调(P<0.01),而间质标志物Vimentin表达上调(图5B)。划痕损伤修复和Transwell迁移实验结果显示,PGC-1α敲低增加了细胞损伤修复能力(图5C-D)。CCK-8实验结果显示PGC-1α敲低并未明显改变细胞的生长速率(图5E),因此可排除细胞增殖导致的可能性。PGC-1α敲低除了可以促进细胞损伤修复能力之外,对肿瘤细胞的干性能力也起到了促进作用(图5F)。综上所述,TGF-β通过经典的Smad通路抑制PGC-1α的转录表达,进而影响线粒体的功能,癌细胞为了改善生存条件及获取更为充分的营养,一方面趋向于向干细胞样转化,另一方面也趋向于EMT途径。

A:评估构建的 A549-tet-shPGC-1α细胞中PGC-1α的敲低效率;B:A549-tet-shPGC-1α细胞中E-cadherin、Vimentin和β-actin的检测及定量结果;C:划痕试验的代表性图像和定量结果(×100);D:Transwell 迁移分析的代表性图像和定量结果 (×100);E:CCK-8实验结果;F:Sphere分析的代表性图像和定量结果 (×100)。1) P<0.05
3 讨论
本研究发现TGF-β可通过经典的Smad通路改变肺癌细胞线粒体的表型与功能。进一步的机制探索发现,TGF-β调控负责线粒体生物合成及代谢的转录共调控因子PGC-1α的表达,改变线粒体的表型和功能。反过来,线粒体的改变促进肺癌细胞EMT表型的增加。
EMT是发育的逆分化过程,即在EMT期间,通过相关的金属基质酶将细胞-细胞连续嵌入的非运动、极化上皮细胞形成的细胞集合溶解并转化为单个的、非极化、运动和侵入性间充质细胞[20-22]。在病理情况下,如肿瘤中EMT可使分化细胞向干细胞样细胞转变,促进癌细胞迁移、侵袭、转移和耐药[10-11]。TGF-β是其中重要的调控因子之一,目前已报道TGF-β在多种癌症中通过EMT促进肿瘤的恶性进展,包括乳腺癌、肝癌、胰腺癌及肺癌等[23-27]。TGF-β与其受体TGFBR1/2结合,主要通过经典Smad和非经典Smad信号通路调控细胞EMT[11]。有研究报道FOXA1和PGC-1α响应TGF-β参与EMT[28]及PGC-1α介导脂肪酸氧化促进鼻咽癌细胞EMT[29],但其并未详细研究肺癌患者线粒体功能在此过程中扮演什么角色。作为细胞的能量工厂,线粒体的表型和功能改变与干细胞分化或成体细胞逆分化有着密切关系[16]。此过程涉及多个生物进程的调控,仍有许多未知的新机制有待阐明。本研究首先验证TGF-β对线粒体表型和功能的影响。利用TGF-β诱导肺癌A549细胞EMT的模型,通过流式检测评价线粒体膜电位和ROS水平的变化,结果显示TGF-β显著地促进线粒体膜电位和ROS的增加[19]。RT-qPCR检测发现线粒体生成和氧化磷酸化功能相关基因在TGF-β处理后显著下调,这些表型改变提示TGF-β抑制线粒体的功能。进一步通过TGF-β受体抑制剂和Smad2可诱导敲低的方式抑制TGF-β信号激活,可以显著恢复线粒体功能和线粒体相关基因的表达。这些结果表明TGF-β对线粒体的作用是通过经典的Smad信号通路调控的。
代谢重编程是癌细胞的特征,线粒体的功能和代谢网络发生改变,细胞ATP的合成方式由氧化磷酸化转变成以有氧糖酵解途径为主,即所谓的“瓦勃效应”,从而也增加营养摄取和生物原料合成,并反馈影响细胞转录和翻译,促进肿瘤生长和恶性发展[30-31]。PGC-1α是线粒体生成和代谢调控的共转录因子,PGC-1α的表达促进细胞的代谢功能,抑制癌细胞的发生发展[29,32]。在本研究中,为探讨TGF-β对线粒体的作用,利用RT-PCR检测TGF-β对PGC-1α表达的影响,结果发现TGF-β可显著抑制PGC-1α的mRNA水平。基因结果也显示TGF-β可抑制PGC-1α的转录活性。TGFBR抑制剂可恢复TGF-β对PGC-1α的抑制作用,这与敲低Smad2抑制TGF-β信号通路可恢复线粒体相关基因表达的结果一致。另外,为证明PGC-1α对线粒体代谢功能的影响,构建可诱导PGC-1α过表达的A549细胞系,当PGC-1α过表达,可显著恢复由于TGF-β引起的线粒体代谢相关基因的表达下调。实验表明,TGF-β在诱导A549细胞EMT过程中,通过抑制Smad通路抑制PGC-1α的表达,从而影响线粒体代谢功能。
目前Torrano等[32]发现PGC-1α在多个癌种中表达下调,而且其下调可促进癌细胞的转移,且有研究表明在胸主动脉瘤中TGF-β信号传导参与PGC-1α调节[33]。为验证PGC-1α下调在TGF-β诱导的EMT模型中的作用,本研究利用PGC-1α可诱导敲低的A549细胞系来模拟TGF-β引起的PGC-1α表达下调的效果。当PGC-1α诱导敲低后,细胞的上皮标志物E-cadherin表达下调,而间质标志物Vimentin表达上调。另外,通过细胞的损伤修复和迁移实验证明了PGC-1α的敲低可显著增加细胞的迁移能力,这是癌细胞发生转移的前提。由此表明,PGC-1α下调促进细胞的EMT表型。
综上,本研究的结果表明TGF-β能够抑制线粒体的代谢功能,反过来由于线粒体的代谢功能受到抑制,细胞为了能够获取更多的营养物质及改善生存条件,进而促进了细胞EMT。本研究在细胞水平上揭示了TGF-β信号通过线粒体调控细胞EMT的新机制,为理解肿瘤细胞EMT这种逆分化现象提供了新思路,并为此通路的靶向治疗提供理论基础。
作者贡献声明
郑宗耀:提出研究思路和框架,修改论文;陈智鹏:实验和数据分析指导;曾观娣:设计实验、统计分析数据,撰写论文。
利益冲突声明
本研究未受到企业、公司等第三方资助,不存在潜在利益冲突。
猜你喜欢
中学生物学(2021年8期)2021-11-02
海洋通报(2021年1期)2021-07-23
生物学通报(2021年4期)2021-03-16
中国果业信息(2019年1期)2019-01-05
中国畜牧兽医文摘(2018年6期)2018-07-28
现代园艺(2017年21期)2018-01-03
中国康复理论与实践(2015年10期)2015-12-24
科学启蒙(2015年8期)2015-08-07
医学研究杂志(2015年5期)2015-06-10
现代检验医学杂志(2015年5期)2015-02-06
