
过渡金属间隙化合物的合成及电催化性能研究进展
2023-05-17 02:58:34张晓凤徐伟杰向桂姣肖路生黄秋锋
福建师范大学学报(自然科学版) 2023年3期
王 婷,张晓凤,徐伟杰,向桂姣,肖路生,林 深,黄秋锋
(福建师范大学化学与材料学院,福建 福州 350117)
由于全球能源的急剧枯竭和随之而来的环境问题,研究开发清洁、可持续的能源来代替传统的化石燃料迫在眉睫.燃料电池、电解水设备和金属-空气电池等因其固有的高能量密度、环境友好性以及能量转换的安全性而备受关注[1-3].这些设备的性能主要受两方面因素的影响,一方面,它与几个基本电化学过程有关,如析氢反应(hydrogen evolution reaction,HER)、析氧反应(oxygen evolution reaction,OER)和氧还原反应(oxygen reduction reaction,ORR).OER/ORR在空气阴极上的固有动力学迟滞可能导致电池过电位大、实际能量密度有限,HER/OER上也存在类似的动力学迟滞,导致界面电荷转移效率低,即电极的反应速度减慢,进而影响电池的工作效率.另一方面,这些设备的电极由活性电催化剂和支撑基体组成,活性电催化剂是影响其性能的关键因素.但是由于贵金属催化剂如Pt基、Pd基催化剂(ORR、HER),Ru基、Ir基催化剂(OER)等的储量较小,制备成本较高,稳定性差等问题,严重阻碍了现阶段大规模生产和应用[4-5].因此,设计制备具有高效催化活性及稳定性的非贵金属材料来代替贵金属基材料意义重大.
近年来,ORR、OER和HER以及直接液体燃料电池阳极的催化剂,如合金材料[6-9]、碳材料[10-14]和过渡金属材料[15-20]等被广泛研究.尤其是过渡金属材料因其资源丰富,成本低廉而备受关注.过渡金属化合物包括配位化合物和间隙化合物,利用异原子(B、N、P、S等半径较小的原子)插入金属晶格,构建过渡金属间隙化合物,可以使催化剂表面的活性位点大面积暴露,可进一步提高电催化活性[21-23].
过渡金属间隙化合物是一类将碳、氮、磷、硫等原子插入过渡金属晶格并形成化学键的化合物[24-30].纳米结构的TMICs比表面积较大,使其反应物、电解质和活性材料之间具有足够的接触面积.这些优良的特性赋予了TMICs更高的电催化性能,同时极大地提高了它的应用价值,特别是在有关能量转换的催化方面,在很大程度上可替代Pt族类贵金属,在ORR、HER以及OER方面有了实质性的突破.同时,在制备具有增强电化学性能的纳米结构TMICs方面也已经取得了很大进展[31-37].
1 过渡金属间隙化合物的结构与特性
过渡金属间隙化合物通常有面心立方结构(fcc)、六边形封闭填充结构(hcp)和简单六边形结构(hex).根据Hägg规则,TMICs的晶体结构由原子半径比rx/rm决定,其中rx表示非金属原子的半径,rm表示金属原子的半径.如果rx/rm之比在0.41~0.59范围内,非金属原子占据金属晶格的最大间隙位置,形成简单结构,如fcc、hcp和hex;如果rx/rm之比大于0.59时,则形成结构复杂的间隙相[24-25,38].同时TMICs具有金属键、共价键和离子键3种键型,其金属特性表现出高导电性,共价键赋予其高硬度、低脆性以及更好的应力耐受性.同时,由于金属原子和非金属原子之间的相互作用,离子键显示出类似贵金属的电子结构,表现出类似Pt的电子性质以及潜在的类Pt电催化行为[39].以过渡金属碳化物为例,通过将碳原子引入过渡金属晶格制备出相应的碳化物.碳原子优先选择占据母体金属的最大位置,碳化物的最终结构由几何和电子因素决定.同时,电子因素可以用金属的Engel-Brewer理论解释,它通过碳的s-p轨道与金属原子的s-p-d带相互作用导致间隙化合物的形成.晶格碳原子又延长了金属-金属键的距离,从而通过电子从金属原子转移到碳原子上,改变了d带电子密度,导致其与金属类似物不同的吸附/解吸特性[40-41].此外,TMICs具有广阔的电化学活性面积和丰富的表面缺陷,可作为载体来锚定单个金属中心.通过调整TMICs的形貌、电荷转移能力、电子结构和表面缺陷,可以为优化TMICs负载单原子位催化剂的催化活性提供一条可观途径.同时,也为作为直接液体燃料电池阳极催化剂奠定基础.TMICs还具有较高的导电性以及良好的pH耐受性等特点,呈现出超高的热稳定性,以及优异的抗中毒和耐酸碱腐蚀能力,在电催化领域具有极大的开发潜力,可择优应用于ORR、HER以及OER中.
2 过渡金属间隙化合物的合成
许多合成路线或制备方法均可获得过渡金属间隙化合物,主要合成方法涉及金属前体与TMICs的C源、N源、P源以及S源,分别在高达2 000 ℃的高温下反应.虽然这些方法很难控制产品形态和其他杂质聚集,但依旧是研究者用于合成各种TMICs的首选路线[42-44].本节主要以程序升温还原法、热解还原法、固相反应法、有机-无机混合物法等方法分别进行了讨论.
2.1 程序升温还原法
程序升温渗碳法是以过渡金属氧化物作为前体,流动碳氢化合物作为碳源,随着程序升温,生成所需的过渡金属碳化物(TMC)[45]的方法.至今为止该方法仍然是制备TMC最广泛的通用合成方法.渗碳过程中的碳原子来自高温下碳氢气体的分解,因此碳前驱体的形态对碳前驱体的种类高度敏感,其中最常用的气态碳源是甲烷和氢气、乙烷和氢气、丁烷和氢气等混合物[46].同时,TMC的电子结构和化学吸附高度依赖于其暴露的单晶表面.为了制备具有高HER活性的单晶TMC,如图1所示,Xu等[47]提出了一种单晶V8C7合成策略,即通过双石墨化碳涂层来保持V8C7的结构稳定性,从而调节V8C7的晶体生长速率,最终形成泡沫镍表面上石墨包覆的V8C7纳米片(V8C7@GC NSs/NF).
程序升温还原法因其合成工艺简单而广泛应用于TMICs的制备.通过控制碳源等和金属前驱体的比例以及反应条件,可达到调节晶体结构、形貌、孔隙率和比表面积的目的.然而,由于碳氢化合物的分解,这种方法制备的TMC通常受到聚合碳的污染,大大减少了活性中心的数量,并且难以完全去除.
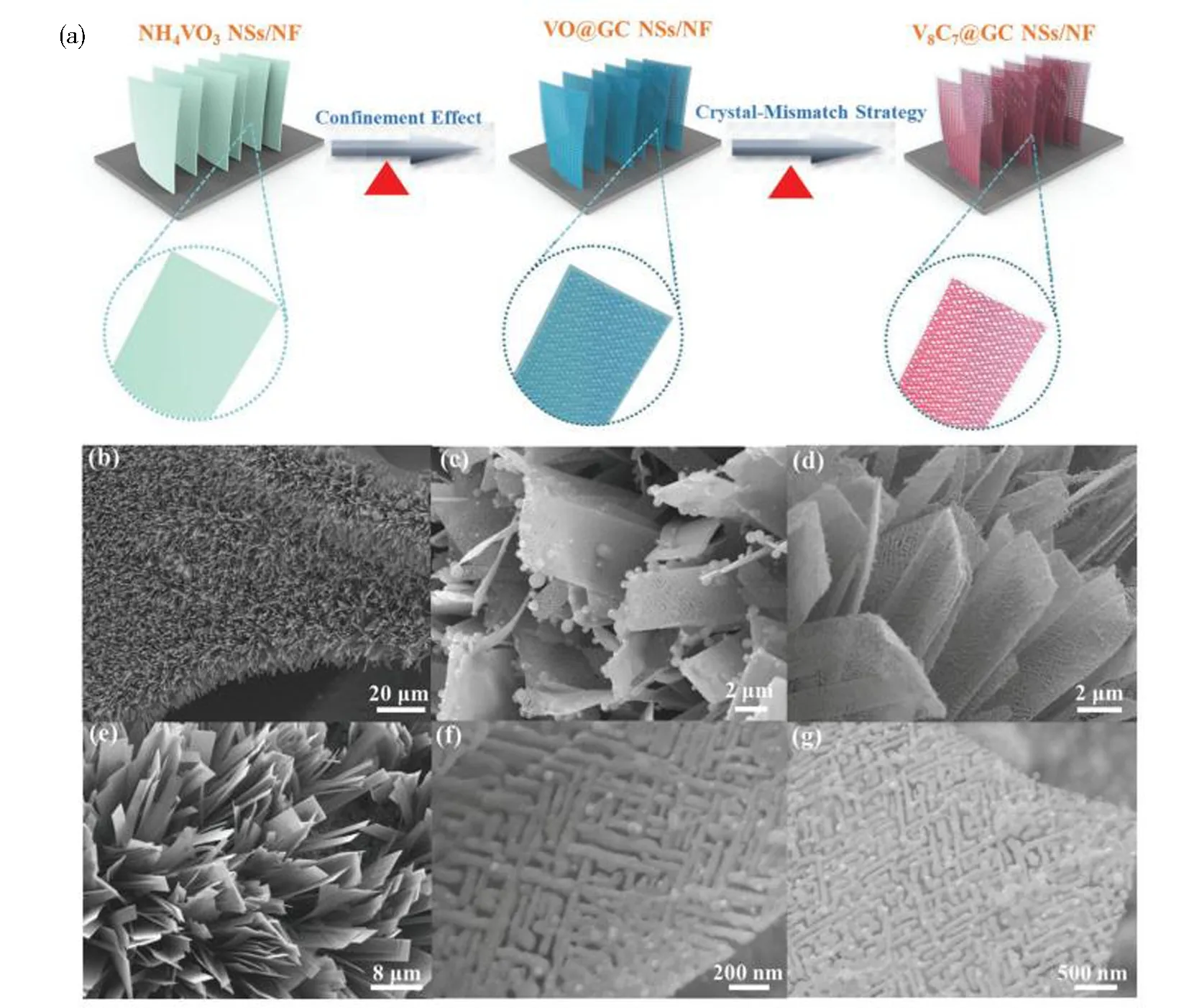
(a)V8C7@GC NSs/NF制备流程图;(b)和(e)分别为NH4VO3 NSs/NF低倍和高倍FE-SEM图;(c)和(f) 分别为VO@GC NSs/NF低倍和高倍FE-SEM图;(d)和(g)分别为V8C7@GC NSs/NF低倍和高倍FE-SEM图图1 V8C7@GC NSs/NF的制备及形貌[47]Fig.1 Preparation and morphology of V8C7@GC NSs/NF[47]
2.2 热解还原法
热解还原法被认为是合成TMICs的基本方法之一,其中金属或合适的金属前体(如金属氧化物)与合适的碳源、氮源或磷源直接反应,如尿素、二氰二胺或磷酸盐等,可以合成相应的碳化物、氮化物或磷化物.基于这一策略,Kim等[48]合成了分散良好的Mo2C、Fe3C和WC·W2C纳米粒子,这些纳米粒子由还原石墨氧化物(rGO)支撑(图2).所得碳化物的粒径小于其氧化物前体的粒径,表明rGO基质在碳热氢反应过程中通过重建粒子来减小粒径方面起着关键作用.热解还原法避免了含碳气体的使用,与含碳气体程序升温还原法相比,该方法制备的碳化物一定程度上降低了表面聚合物的含碳量.然而,反应过程仍会释放出碳、氮、磷等物种,不可避免地使产品表面受到聚合碳、氮、磷等的轻微污染[49].
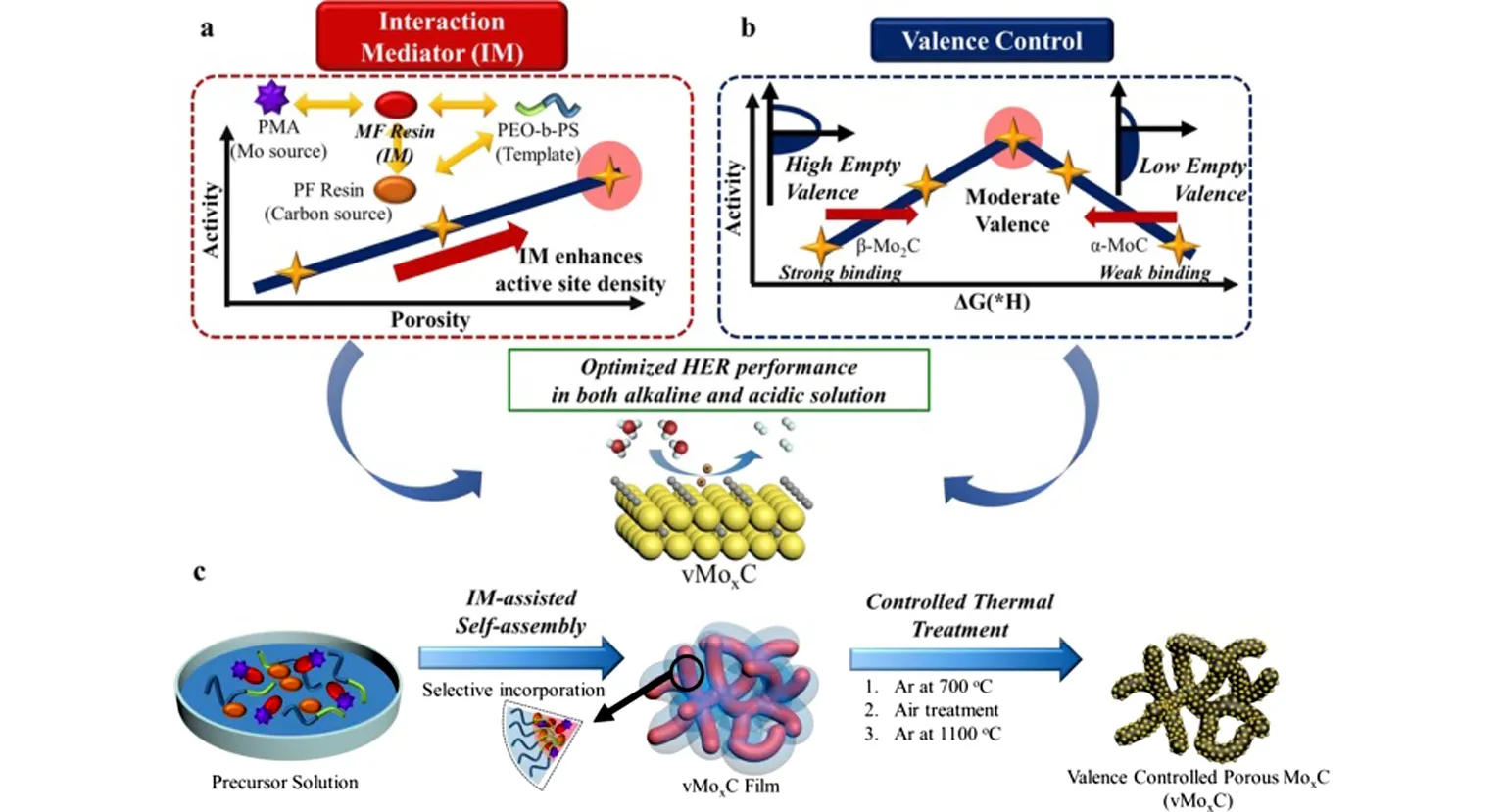
(a)相互作用介质;(b)MoxC价控;(c)vMoxC合成流程图[48]图2 IM辅助价控MoxC(vMoxC)合成示意图Fig.2 Schematic representation of IM-assisted synthesis of valence-controlled MoxC (vMoxC)[48]
2.3 固相反应法
固相反应是一种涉及高温固体反应的合成方法,即将金属盐与碳源、磷源等混合,然后在400~1 000 ℃惰性气氛或氢气气氛中进行程序升温还原[50].如图3a所示,Mu等[51]利用玉米秸秆作为碳前体与四水钼酸铵混合,在900 ℃Ar气氛中反应,制备了嵌入多孔碳纳米片中的Mo2C纳米颗粒(Mo2C NPs).其中玉米秆提供了丰富的多孔通道,有利于其与电解质接触,碳基质也可以抑制Mo2C纳米粒子的聚集,从而增加暴露的活性位点数量.该复合材料表现出良好的HER电催化活性,在0.5 mol·L-1H2SO4中,电流密度达到10 mA·cm-2时,它的过电位为114 mV,Tafel斜率为52 mV·dec-1(图3b、3c).
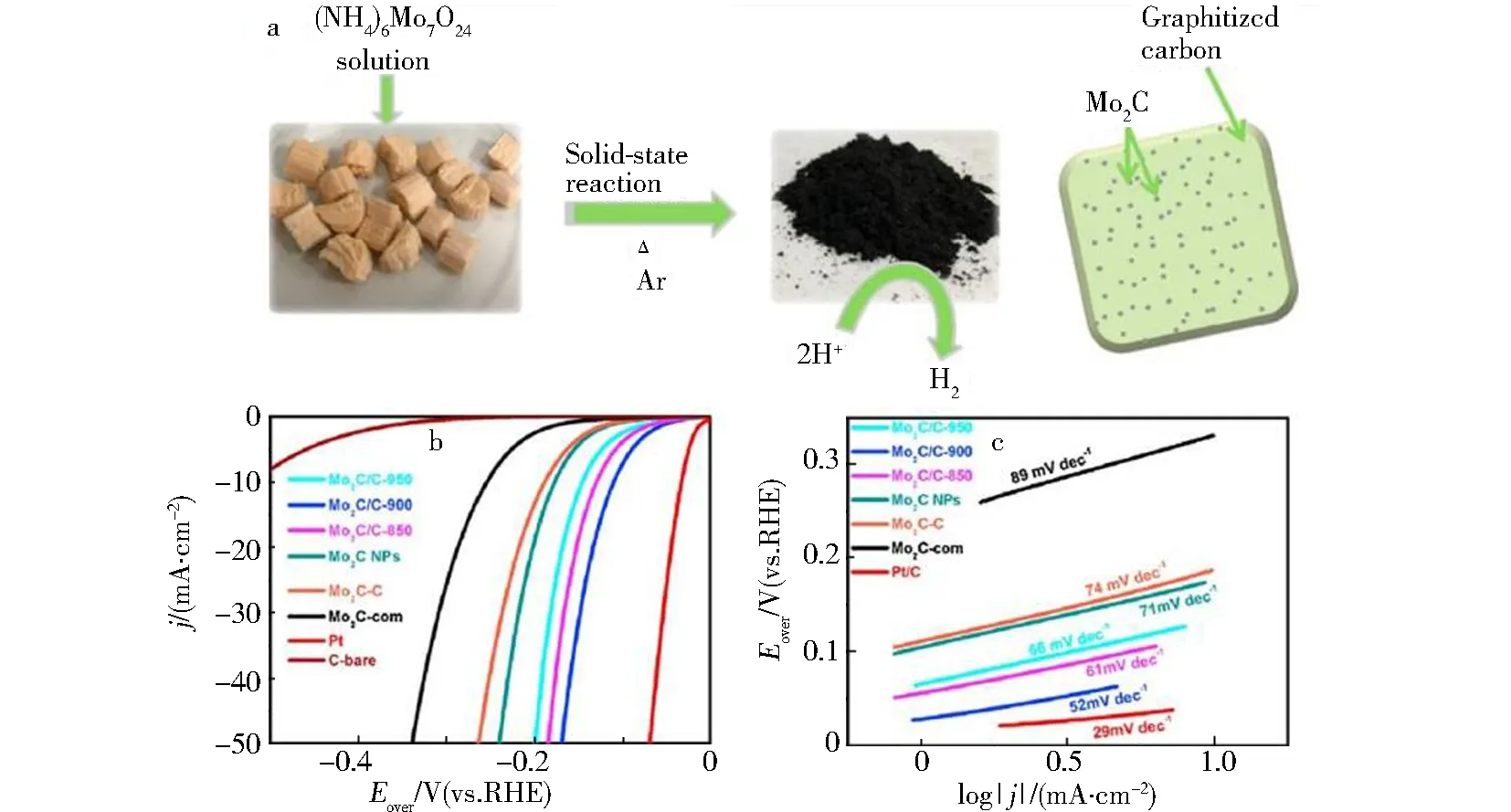
(a)Mo2C/C制备流程图;(b)1 mV·s-1扫速下0.5 mol·L-1 H2SO4中的Mo2C/C-850、Mo2C/C-900、Mo2C/C-950、Mo2CNP、Mo2C-C、Mo2C-com和Pt/C的极化曲线和(c)Tafel曲线图图3 Mo2C/C 的制备及其HER性能[51]Fig.3 Preparation and HER performance of Mo2C/C [51]
2.4 有机-无机混合物法
有机-无机杂化方法是制备TMICs的一种简便方法,即通过金属前体与相应有机物反应制备杂化复合材料,然后在惰性气氛中通过复杂前体的相对低温热分解生成TMICs[52].金属-有机骨架(MOF)是由金属离子和有机配体生成的高比表面积的多孔材料,因其具有高的孔隙率和好的化学稳定性,MOF比其他多孔材料具有更广泛的应用前景.同样,这种有机-无机杂化特性使其成为制备多孔TMICs的潜在前驱体[53-55].Ding等[56]选择了3种不同组分的铁基普鲁士蓝类似物(PBA)作为前驱体,通过简单的热解处理将其转化为相应的金属磷化物,通过调节磷化温度,得到一系列FeCoP、FeNiP和FeMnP,其中FeCoP在400 ℃磷化温度下具有最明显的多孔结构和最宽的孔径分布,在OER中显示出优异的催化活性以及出色的稳定性.如图4a所示,Shi等[57]通过引入MOF(Mo3(BTC)2)作为混合前体制备了高度分散的石墨包覆的纳米MoC(nanoMoC@GS)电催化剂,其比表面积高达187 m2·g-1,并且在0.5 mol·L-1H2SO4中表现出良好的HER电催化活性,电流密度为10 mA·cm-2,其过电位为124 mV,Tafel斜率为43 mV·dec-1(图5a).如图4b所示,Yu等[58]以普鲁士蓝类似物(PBA)作为前驱体,通过MOF 辅助合成方法,将无定形碳结合到Ni-P化合物中,合成了Ni-P多孔纳米板.测试发现,Ni-P纳米板表现出更高的电流密度和更负的OER起始电位.
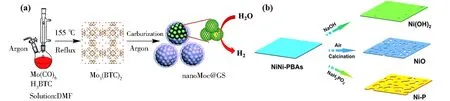
(a)nanoMoC@GS的合成示意图;(b)Ni(OH)2、NiO以及Ni-P的合成示意图图4 有机-无机混合法制备复合物[57-58]Fig.4 Preparation of composite materials by organic-inorganic mixing method[57-58]
3 过渡金属间隙化合物的电催化性能
随着化石燃料的持续消耗,人们对能源危机的认识日益提高,在过去几十年中,人们迫切想要寻找一种清洁能源来解决能源短缺的现实问题.氢能、燃料电池、金属-空气电池等作为有前景的绿色能源,在便携式电子设备和电动汽车等应用中具有巨大潜力.通过电解水生产氢燃料是提供氢能的有效途径.但是由于电解水过程中涉及的OER和HER反应速率常数小,超电势和能量损失较高,因此极大地限制了电解水制氢的发展[59-61].但是研究发现,TMICs导电性良好,结构较稳定,且具有丰富的活性位点,它不但可以代替Pt基、Pd基这些贵金属作为ORR、HER、OER更加普适的催化剂,还在甲酸、甲醇等燃料电池中,具有意义深远的用途[62-63].
3.1 ORR催化剂
近年来,人们对寻找ORR的替代催化剂产生了极大的兴趣.大量研究发现,TMICs在ORR中表现出相对稳定的催化活性,有望替代Pt族贵金属催化剂.Zhang等[64]用热解还原法合成介孔 WC@NC 和均匀的 Fe/Co 碳化物,其结构为石墨层封装的Fe3C、Co3C纳米颗粒和与 N掺杂的石墨碳互连的WC.这种独特的成分赋予了催化剂高ORR活性和稳定性.后来,Wang等[65]首次开发了一种由双氰基酰胺和单宁铁骨架衍生的氮掺杂碳纳米片,构建了一种封装的碳化铁纳米颗粒(Fe3C/N-CNS),由于N的掺杂改变了Fe3C的电子结构,在整个pH范围内表现出与Pt/C催化剂相当甚至更好的ORR催化活性,以及长期的甲醇耐受性和稳定性.近几年,研究发现多金属掺杂可以对催化剂的电子结构进行有效调控,能进一步提升其催化活性.例如,Cui等[66]通过掺杂非均匀原子研究了TiC基无贵金属ORR催化剂.N掺杂导致N/TiC的ORR性能增强,同时证实在TiC粉末中引入金属(如Fe、Co),能进一步提高ORR性能.Zhang等[67]通过一步热解法,在不使用NH3的情况下,成功地合成了Co、N共掺杂多孔氮化钒(VCoN)纳米板,显示出优越的ORR性能,与商用的20%Pt/C相比,具有更好的起始电位(~1.02 V)、半波电位(~0.91 V),并在0.1 mol·L-1KOH中以1 600 r·min-1进行2 000次循环后只有较弱的电位偏移(~5 mV).此外,Liu等[68]通过热解含硫氮配位聚合物及碳获得了具有丰富缺陷的ZnS/C纳米颗粒,在碱性介质中显示出良好的ORR电催化性能.由此可见,TMICs已经在ORR反应中得到了广泛的研究,并且催化活性已经达到了理想状态,其成果丰富.
3.2 HER催化剂
作为前途广阔的HER催化剂,TMICs具有优良的pH耐受性,由于在金属晶格中插入的碳、氮、磷、硫等原子,使其晶格常数扩大,而且由于金属d轨道与插入原子的s和p轨道的杂化,导致金属的d带结构变宽,因此具有了类似贵金属的催化性能.An等[69]使用聚多巴胺生物制备氮-碳包封碳化钼磷化物杂化纳米材料(Mo2C-MoP@CP),具有高效的析氢活性和长久的耐用性.Xu等[70]则使用聚乙烯亚胺(PEI)、金属离子和多金属氧酸盐(POM)(Co9P5W27)通过一步高温煅烧合成Co/WC@NC,在0.5 mol·L-1H2SO4和1.0 mol·L-1KOH中均显示出优异的HER催化活性及稳定性,并且发现Co和WC与多孔结构之间的协同效应可以有效地加速析氢过程.Liu等[71]则通过两步水热反应成功合成Mo掺杂的NiCoP@C微球纳米材料,由于元素掺杂和形貌控制的固有特性以及各种组分之间的多种协同策略,最大限度地暴露活性位点并促进电荷快速转移,使其在碱性条件下,达到10 mA·cm-2电流密度仅需74.6 mV过电位,Tafel斜率仅为54.9 mV·dec-1.此外,Mo-NiCoP@C催化剂在25 h测量期间表现出极好的耐久性.Su等[72]研究发现MoS(001)晶面通过掺杂过渡金属(M)或产生S空位(Sv)进行改性,可以使氢的裂解势能降低0.04~0.96 eV.通过比较发现,H2解离活性遵循以下顺序:M修饰的Sv>未修饰的Sv>M修饰的S位点>M修饰Sv-M和Sv修饰的S位置>M和Sv修改的S位点>Sv修改S位点>未修饰S位点.并且与Mo相比,M的外部电子数量的增加会导致与S结合的未成对电子减少.因此为改进MoS2和其他金属掺杂来提高其催化活性提供了可能.Baek等[73]选择适当的钼前驱体进行高温渗碳,制备出高度有序的中孔钼碳化物,即亚稳fcc a-MoC1-x相(MMC),在碱性介质中HER催化活性显著提高,是迄今为止报道的粉末碳化物中相对较好的一种碱性HER催化剂,并且具有比商用Pt/C(20%)更有利的耐久性及稳定性.Xiong等[74]合成的Mo-Co5.47N/N-CoO,证明了同时从界面工程与元素掺杂两方面来改善氮化钴,可以同时提高在碱性和酸性介质中的HER活性,并且在更高的电流密度下优于商业Pt/C催化性能.因此,表明经过改性的氮化钴催化剂具有满足商业标准的潜力.此外,表1中调查了近几年TMICs在HER中的研究现状,通过在10 mA·cm-2的过电位和Tafel斜率可以看出,这些类型的催化剂也具有令人满意的催化活性.以上所有结果都表明TMICs具有成为更好HER催化剂的潜力.
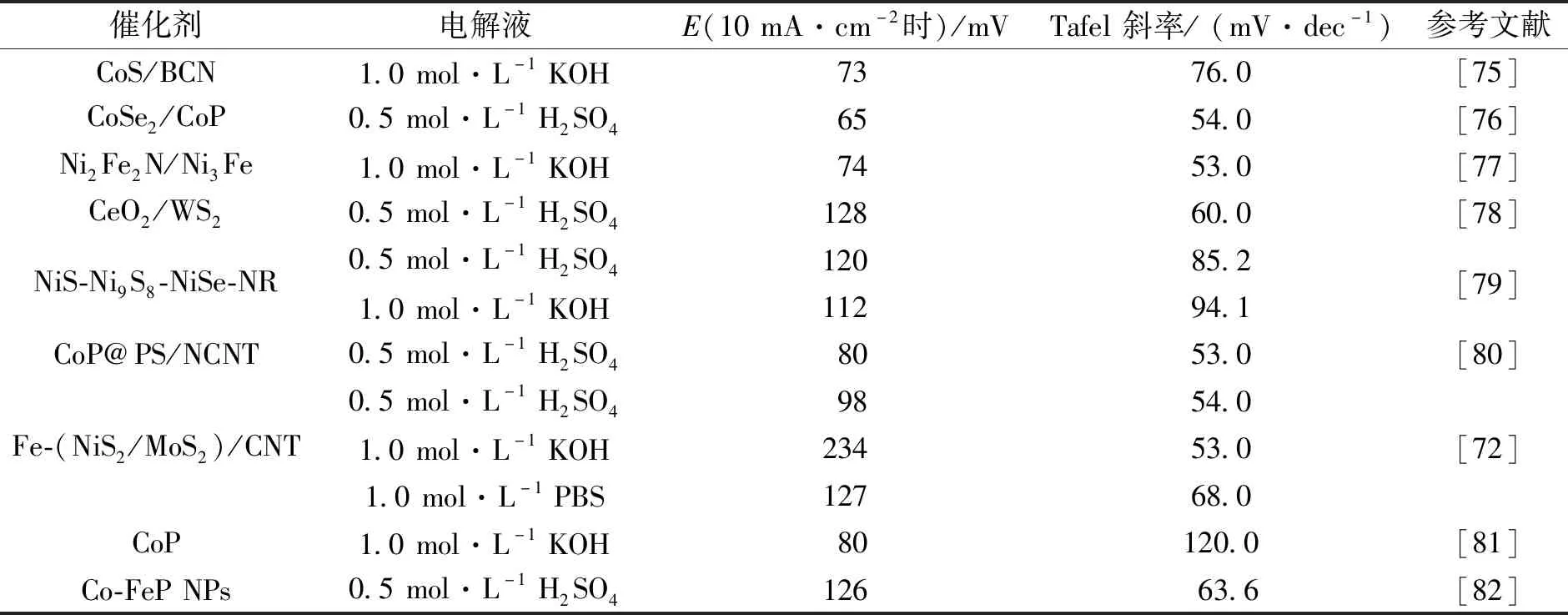
表1 不同物质负载TMICs纳米材料的HER活性比较Tab.1 Comparison of HER activity of TMICs nanomaterials loaded with different substances
3.3 OER催化剂
目前,铱和钌及其氧化物(IrO2和 RuO2)等贵金属催化剂依旧被广泛用于 OER 中.但原材料储量丰度低、成本高以及在碱性介质中的化学稳定性差等特性,严重阻碍了其大规模应用.因此,开发一种更高效、稳定、低成本的 OER 非贵金属电催化剂来替代昂贵的 Ir 和 Ru 基催化剂是发展电催化分解水技术的关键.Ni-Co 基催化剂中氮、磷和硫等极化阴离子的存在可以优化催化剂表面的原子和电子结构,增加活性中心密度和电导率,并提供中等强度的中间体吸附能.因此,Ni-Co 基氮化物、磷化物和硫化物在OER 中得到了广泛和深入的研究.Shanmugam 等[83]采用了一种固态热分解的方法,将不同组分的有机金属前体在其自生压力下在无定形碳中形成分散良好的Co、Ni和Fe磷化物纳米颗粒.嵌入碳基体的不同金属磷化物在1 mol·L-1KOH溶液中表现出显著的OER活性,就Co和Ni磷化物电催化剂而言,当电流密度达到10 mA·cm-2时,所需过电位分别为370和380 mV,与IrO2相比,则具有更小的过电位.Li等[84]制备了铁掺杂的CoO/C(Fe-CoO/C)纳米纤维,在碱性介质中表现出与商业RuO催化剂相当的OER催化活性,当电流密度达到10 mA·cm-2时,其过电位为362 mV.Wang等[85]通过高温热解反应制备了CoNiP2S2,在OER方面表现出优异的性能.Liu等[86]通过将磷化钴与碳化钼(MXene)整合来构建异质结构CoP/Mo2CTx(T是表面端基),具有媲美于RuO2基的OER催化活性,在1 mol·L-1KOH中,10 mA·cm-2处的过电位非常小,为260 mV.此外,表2对比了近几年的OER催化剂的催化活性,发现大部分的Co基和Ni基与N、P、S形成的间隙化合物的OER催化活性是令人满意的.因此,这些催化剂完全可以替代Ir类和Ru类贵金属作为电解水OER的良好催化剂.
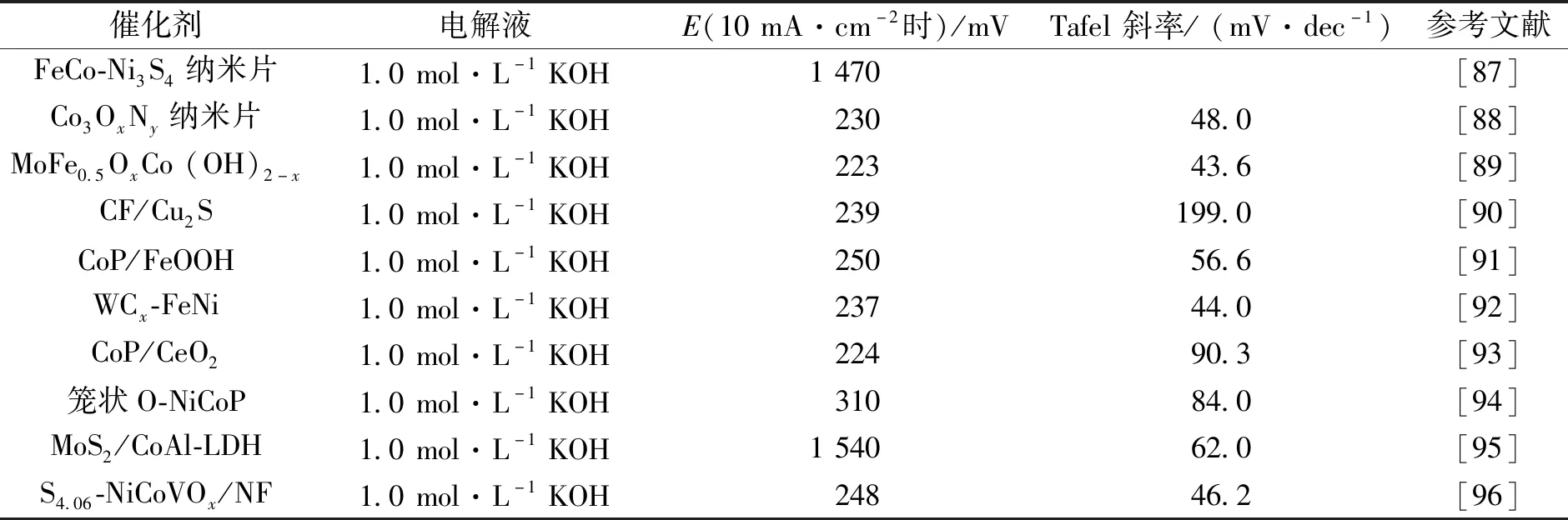
表2 不同物质负载TMICs纳米材料的OER活性比较Tab.2 Comparison of OER activity of TMICs nanomaterials loaded with different substances
3.4 多功能催化剂
过渡金属存在d轨道未完全填充的情况,所以过渡金属可以很容易地提供和获得电子,从而提供相对丰富的电催化反应.尤其是,Ⅷ B的Co基和Ni基化合物在电催化方面得到了广泛的研究扩展.通过调整 Ni/Co基电催化剂的微观形貌、晶体结构、成分和氧化态、异质结构以及与导电材料复合等策略可以有效改善电催化性能.Li等[97]采用一步法合成了铁掺杂多孔磷化钴多面体,具有较低的起始过电位、较大的电流密度、较小的Tafel斜率和良好的电化学稳定性,作为一种高效的HER和OER双功能电催化剂,显著增强了HER和OER双功能电催化活性,在10 mA·cm-2时,过电位分别为116、289 mV,且稳定性良好.同样,Xu等[98]通过简单的磷化处理合成了网状铁掺杂的CoP多孔纳米片(Fe-CoP-HNSs),可作为HER和OER的高效双功能电催化剂,达到10 mA·cm-2时的过电位分别为79、220 mV.该研究为设计具有多孔纳米片结构的过渡金属化合物电催化剂用于可再生能源生产提供了线索.此外,Li等[99]通过热解还原法在泡沫镍上制备了自支撑的掺铁Ni2P纳米片阵列.优化后的Fe掺杂Ni2P [(Ni0.33Fe0.67)2P]表现出优异的HER活性和OER活性,当电流密度达到50 mA·cm-2时,其过电位分别为214 和230 mV,均优于商业Ir/C.除了过渡金属磷化物外,过渡金属硫化物同样也具有双功能的催化活性.Tong等[100]将N-掺杂CoS2纳米颗粒构建在N-S共掺杂石墨烯纳米片上,制备了一种N-CoS2/G催化剂,该催化剂具有良好的HER和OER双功能活性.在电流密度为10 mA·cm-2时,N-CoS2/G对OER的过电位为260 mV,对HER的过电位为109 mV,表现出了优越的催化活性.Wan等[101]合成高度有序的介孔NiSx/NMC(NMC表示氮掺杂介孔碳)纳米杂化物,在0.1 mol·L-1KOH中,达到10 mA·cm-2时,ORR的过电位为0.89 V,OER的过电位为1.57 V.并且在锌-空气电池中使用这种电催化剂,最大功率密度高达186 mW·cm-2,同时具有805 Wh·kg-1的能量密度,以及300次充放电循环100 h的长期循环耐久性.Xu等[102]制备了一种氮掺杂碳负载金属焦磷酸盐(M2P2O7@NC)催化剂,其中M = Co或Zn.由于该催化剂具有大量的活性中心、良好的碳网络导电性以及M2P2O7纳米颗粒与N掺杂碳层之间的协同效应,极大地提高了对ORR和HER的催化性能.在碱性介质中,ORR整个电位范围内M2P2O7@NC的半波电位为0.793 V,接近Pt/C的0.830 V.并且扩散极限电流密度为6.100 mA·cm-2,优于Pt/C的5.427 mA·cm-2;在0.5 mol·L-1H2SO4中进行HER时,电流密度为10 mA·cm-2时的过电位为180 mV.Cai等[15]利用水热及高温煅烧法合成了具有异质结构的MoS2‖CoP/NPC(NPC为N、P共掺杂碳)催化剂,MoS2‖CoP/NPC催化OER和HER方面先显示了低的过电位(306 mV,149 mV),催化ORR则具有较正的半波电位(0.790 V),表明MoS2‖CoP/NPC分别对OER、HER、ORR具有高效的电催化性能.此外,在锌空气组装试验中,MoS2‖CoP/NPC作为电催化剂具有较高的比容量(775.65 mAh·g-1).Surendran等[103]开发了碳纳米纤维包覆的NiCoP纳米颗粒(NiCoP/CNF),在碱性条件下(KOH溶液),可作为ORR、OER和HER的电催化剂,NiCoP/CNF作为ORR催化剂,半波电位为0.82 V,并且对于OER和HER,在10 mA·cm-2的电流密度下,分别具有相对较低的过电位(268、130 mV).这些研究成果均为开发新型双功能电解水催化剂提供了新思路,且对于能量转换应用,具有高效性、高活性和长期稳定性.
3.5 直接燃料电池阳极催化剂
直接液体燃料电池因具有燃料来源广泛、成本低、不存在严重的环境污染等特点受到广泛关注,特别是直接甲醇/甲酸燃料电池作为许多设备的替代电源引起了越来越多的兴趣,例如汽车和便携式电话等便携式电子设备[104-106].然而,降低成本是许多研究领域迫切需要解决的问题.因此,减少贵金属催化剂的用量或提高其催化效率是推进其商业化的重要手段[107].Cao等[108]通过乙二醇还原成功合成了Pt-Ni2P/GO催化剂,物理和化学测试表明,Ni2P的加入很大程度上提高了甲醇氧化的电催化活性和稳定性,这可归因于Pt和Ni2P之间的界面的协同作用以及反应中间体扩散产生的溢出效应.这为降低甲醇燃料电池的成本提供了新思路.Abbas等[109]制备了碳纳米纤维负载镍锰碳化物(NiCeMnC@CNFs)催化剂,在碱性条件下表现出优异的甲醇电催化性能,其氧化电流密度可达110.34 mA·cm-2.Xu等[110]首次报道了通过多步反应合成具有明确八面体形状的空心磷化钴八面体纳米粒子(CoP OCHs),在OER中表现出高的表观催化活性,在240 mV的过电位下便能达到10 mA·cm-2的电流密度.同时,对甲醇氧化反应(MOR)的高电催化活性,优于迄今报道的大多数基于金属磷化物的MOR催化剂.Wei等[111]通过简便的水热法和低温磷化工艺合成了双金属FeNi2P/C杂化物,在酸性溶液中甲醇氧化过程中表现出1 125 mA·mg-1的优异催化活性.Liu等[112]成功制备的硼-氮官能化的碳纳米管(CNT)负载的PdMoP0.01/OB-CNT-N催化剂表现出比商业Pd/C更出色的甲酸氧化催化活性和稳定性,其质量活性(2 176 mA·mg-1)和稳定性分别比商业Pd/C高7和8.7倍.催化剂电催化性能的显著提高主要是由于引入了Mo和P,这改变了Pd的电子结构,并产生了Pd和Pd-Mo的双活性位点,从而增加了有效反应中心.Wang等[63]通过热解磷化处理,制备了多孔磷化钯纳米管 (PdxPy-PNTs),合成的Pd3P-PNTs具有多孔纳米管形貌,有利于暴露活性位点,加快产物/反应物的传质速率;丰富的缺陷原子和优异的自稳定性,提高了催化剂的电催活性和甲酸氧化耐久性.此外,P的引入降低了COads的吸附能,促进了高活性Pd位点的持续暴露.同时,P的电子吸附作用使Pd处于缺电子状态,从而抑制了甲酸分解反应的发生.这项研究为设计具有高电催化活性、耐久性和甲酸氧化选择性的1D Pd基纳米材料提供了一种新的策略,并证明Pd3P-PNT有潜力成为直接甲酸燃料电池的有效阳极电催化剂.虽然对于直接液体燃料电池阳极催化剂来说,Pt、Pd等贵金属类催化剂依旧是最有效的催化剂,但是提高铂贵金属的利用率和电催化性能被公认是直接液体燃料电池商业化最亟需解决的关键问题.
4 小结与展望
在过去的几年中,各种过渡金属间隙化合物(如氮化物、碳化物、磷化物、硫化物等)因其固有的金属性质和新颖的性能,包括类Pt的电子结构,优异的导电性、催化活性、稳定性以及原料的经济性,制备的简易性等,在电催化领域成为代替Pt族贵金属有潜力的候选材料,有望被广泛用作电化学能量应用的电极材料.在这篇综述中,全面总结了 TMICs 的最新进展,包括结构性质、制备方法以及电化学能量转换应用.
首先,本文系统地介绍了TMICs的电子结构,不同杂原子掺杂的协同作用,使过渡金属基纳米催化剂的电子结构得到有效调节,为提高催化剂的催化活性提供了新思路.同时,对多功能催化剂的制备和发展具有重要意义.其次,总结了TMICs的制备方法,选择合适的制备途径不仅可以有效地控制催化剂的形貌,还可以更大程度地暴露其活性位点,使其催化活性进一步得到提高.最后,总结了近几年TMICs在能量转换器件上的应用.
尽管 TMICs在电化学能量转换的应用中显示出许多优势,并且在过去几年中发展迅速,但仍然存在一些问题和挑战需要不断探索和改进.(1)在合成方面存在一定的局限性,由于其合成往往需要高温热解,这使得碳、氮等物质聚集,导致负载不均匀,一定程度上堵塞了催化活性位点,降低了催化效率.这就需要不断地对过渡金属间隙化合物进行改性,通过控制过渡金属间隙化合物的合成方法,其形貌、尺寸、多孔结构、孔隙大小和缺陷状态均被合理地调控,从而提高其催化活性.(2)应用面不够宽泛,很多催化剂在酸性介质中能表现出优良的催化活性,但在碱性介质中的催化活性会很大程度上降低甚至不耐碱.因此,通过研究开发在较宽pH值范围电解液中具有良好催化活性的催化剂,进一步拓宽其应用领域是相当有必要的.(3)催化功能较为单一,一定程度上限制了其应用,因此开发具有多功能的催化剂也亟需解决.
猜你喜欢
物理化学学报(2024年9期)2024-09-27 00:00:00
电镀与精饰(2022年10期)2022-10-14 08:37:12
中国有色金属学报(2018年2期)2018-03-26 07:58:37
电镀与环保(2017年6期)2018-01-30 08:33:37
电镀与环保(2017年5期)2017-12-19 12:06:05
电镀与环保(2017年3期)2017-06-23 08:24:51
中南大学学报(自然科学版)(2016年2期)2017-01-19 07:37:25
淮南师范学院学报(2015年3期)2015-03-22 01:16:19
河北科技大学学报(2015年5期)2015-03-11 16:16:34
无机化学学报(2014年4期)2014-02-28 17:31:23
