
Disease-associated oligodendrocyte signatures in neurodegenerative disease: the known and unknown
2023-03-23 08:13KristenSchusterHayleyMcLoughlin
中国神经再生研究(英文版) 2023年10期
Kristen H.Schuster, Hayley S.McLoughlin
Oligodendrocytes are one of the most abundant cell types in the central nervous system (CNS) and act in close contact with neurons to assist with their cellular function and health (Kuhn et al., 2019).Oligodendrocytes play particular roles in rapid nerve impulse conduction through the wrapping of their myelinating projections around a nerve axon, as well as by offering trophic and metabolic support for the high energy expenditure of neurons.Additionally,oligodendrocytes have been found to regulate axonal health directly or indirectly by monitoring immune networks between glia and neurons.Unlike neurons, apoptotic mature oligodendrocytes can be replaced via differentiation from a pool of oligodendrocyte precursor cells (OPCs).Proliferative OPCs remain within the CNS throughout adulthood(Yalcin and Monje, 2021) and function as more than just a progenitor pool to replace lost myelinating oligodendrocytes.OPCs have been shown to interact with other oligodendrocytes, neurons, astrocytes,and microglia, and have roles in myelin maintenance,synaptic formation, blood-brain barrier support, and immune responses (Yalcin and Monje, 2021).These intercellular interactions, in addition to their capacity to become myelinating oligodendrocytes, could make OPCs an intriguing target for therapeutic intervention in demyelinating diseases, such as multiple sclerosis,and other neurodegenerative disorders with recent reports of white matter abnormalities and oligodendrocyte dysfunction (Philips et al., 2013;Ferrari Bardile et al., 2019; Errea and Rodriguez-Oroz,2021; Kenigsbuch et al., 2022; Schuster et al., 2022).White matter degeneration has recently emerged as a shared pathology among many late-onset neurodegenerative diseases.Another pathological hallmark of these disorders is the intracellular aggregation of misfolded proteins, leading them to be commonly referred to as proteinopathies.These proteinopathy neurodegenerative diseases include Alzheimer’s disease (AD), tauopathies,Parkinson’s disease (PD), amyotrophic lateral sclerosis, and polyglutamine expansion diseases such as spinocerebellar ataxia type 3 (SCA3) and Huntington’s disease (HD).Protein aggregates have primarily been described in neurons within vulnerable brain regions of disease, leading to extensive study of pathomechanisms in affected neuronal populations.However, in the last decade or so, underexplored complex networks of interactions between oligodendrocytes and neurons in many neurodegenerative diseases have emerged.The complex networks of interactions between diseaseassociated oligodendrocytes and neurons lead to many interesting questions.Are disease-associated oligodendrocyte signatures a secondary effect of progressive neuronal degeneration? Could diseaseassociated oligodendrocytes be serving to signal cellular stress prior to overt cell loss of the precious post-mitotic neurons? Here, we present a perspective on the known and unknowns of disease-associated oligodendrocytes in neurodegenerative diseases.We touch briefly on the seminal findings that are leading to the breadth of disease-associated oligodendrocyte signatures across neurodegenerative diseases and highlight the tools that are being employed to elucidate the role of oligodendrocytes in disease(Figure 1).Finally, we cover the challenges towards progression in this developing field of study and proffer future directions to bridge the gaps that inform the role disease-associated oligodendrocytes might play in pathogenetic disease mechanisms, as well as the potential therapeutic opportunities.
Establishing disease-associated oligodendrocytesignatures in human contexts:Because glia,oligodendrocytes in particular, have only recently been emphasized as important contributors to noncanonical demyelinating neurodegenerative diseases,the models and methods to study these diseased cells have been limited.Much of the disease-associated oligodendrocyte research was first documented via histological evaluation of postmortem patient tissue or RNA sequencing of animal models of disease(Figure 1c).Importantly, these results are now being corroborated by longitudinal imaging of white matter changes in patients, as well as through biochemical,histological, and functional oligodendrocyte studies in animal models of disease (Figure 1a–e).However,critical advancements in this field will depend upon the development and implementation of tools that mechanistically perturb and assess oligodendrocyte function both normally and in disease.
Innovations in human studies of white matter abnormalities in neurodegenerative disease will be crucial to maintain the relevance of animal models.Postmortem patient tissue analysis provides interesting late-stage snapshots of brain pathology,but it does little to tease apart whether diseaseassociated oligodendrocyte signatures are a driver of disease or just a secondary consequence of primary pathology.Rather, for longitudinal assessment in human disease, non-invasive white matter imaging can inform the progression of oligodendrocyte-rich white matter abnormalities (Figure 1e).For example,longitudinal diffusor tensor imaging in patients can provide substantial insight of region-dependent white matter loss.
To fully understand the loss of brain white matter in neurodegenerative patients, biochemical,histological, and functional studies can be carried out in both cellular and animal models.The use of primary oligodendrocyte cell culture from disease models have shown benefit in determining the cellautonomous oligodendrocyte processes disrupted in a neurodegenerative disease (Kenigsbuch et al.,2022; Schuster et al., 2022;Figure 1f).However,access to human primary oligodendrocytes impedes the translation of findings from mouse disease models.One work around to this problem is induced pluripotent stem cell reprogramming, where the induction of master oligodendrocyte transcription factors can reprogram induced pluripotent stem cells into oligodendrocytes (Ng et al., 2021) or brain organoids (Figure 1gandh).Therefore, stem cells could provide an efficient and standardized tool for the generation of human oligodendrocytes from an expandable source.
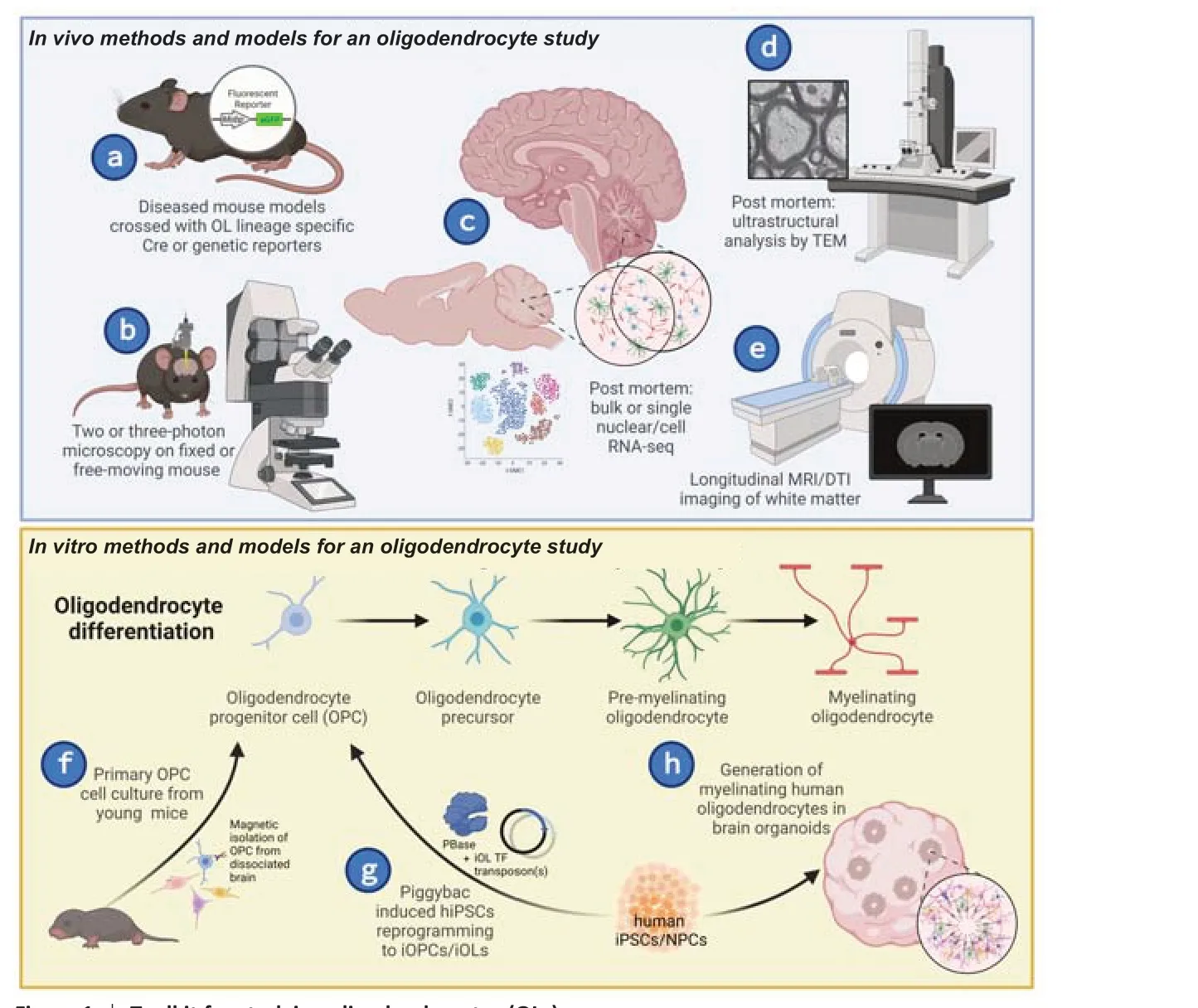
Figure 1|Toolkit for studying oligodendrocytes (OLs).
Utility of animal and cellular models to study oligodendrocyte dysfunction in disease:In vivodisease models are excellent for mechanistic insight and proof of concept studies, however, in polygenic risk disorders, such as AD or PD, it can be difficult to tease apart the direct pathogenic mechanisms.Instead, monogenetic disorders, such as HD and SCA3,could stand in as paradigmatic neurodegenerative diseases to inform on pathomechanisms of oligodendrocyte impairment.An illustrative example of this comes from the HD field.Utilizing conditional oligodendrocyte-specific mouse models (Figure 1a),Huang et al.(2015) demonstrated that overexpression of mutant Huntingtin, the disease gene, specifically within mature myelinating oligodendrocyte cells resulted in cell-autonomous contributions to pathological and behavioral HD pathogenesis.Conversely, the conditional genetic knockout of mutant Huntingtin disease gene expression in oligodendrocyte lineages alone was sufficient to remediate aspects of HD progression (Ferrari Bardile et al., 2019).Similar work in another polyglutamine mouse model, SCA3, utilized a SCA3 mouse model crossed with a mature oligodendrocyte genetic reporter line (Figure 1a) to visualize oligodendrocyte maturation impairments that progressed in a regionally and temporally vulnerable manner.This maturation impairment was further determined to be a cell-autonomous effect through primary oligodendrocyte cell culture techniques (Schuster et al., 2022).Because of the monogenetic inheritance of these CAG repeat expansion disorders, the diseasespecific pathomechanisms underlying this cellautonomous oligodendrocyte impairment can be interrogated using both mouse and cellular models.Such studies can inform hypotheses to be tested in other neurodegenerative disease models.These studies highlight the power ofin vivoandin vitroexploration of disease-associated oligodendrocyte dysfunction in model systems.
Toward animal model studies in the more common neurodegenerative diseases, a recently published study by Kenigsbuch et al.(2022) highlighted across multiple AD mouse models an impairment of oligodendrocyte maturation that increased in association with brain pathology, such as amyloid-beta plaque and tau accumulation.This work suggested disease-associated oligodendrocyte signatures as a common response to severe pathological conditions in AD.Additionally, reports from PD resolved through RNA sequencing found oligodendrocytes as a novel contributor to the etiology of the disease (Errea and Rodriguez-Oroz, 2021).Notably, in AD and PD, the mechanism and impact of these disease-associated oligodendrocytes are not yet clear.Specifically,whether oligodendrocyte maturation dysfunction is either the cause or the consequence of neuronal degeneration across these proteinopathies must be further investigated.
Mechanistic insights from common oligodendrocytesignatures in disease and challenges to overcome:Of particular relevance to age-related neurodegenerative disease, the oligodendrocyte field is exploring the mechanisms regulating oligodendrocyte maturation state throughout the aging process.Among these are common cellular mechanisms that are ratelimiting building blocks, such as lipid biosynthesis and myelin protein translation, yet are essential for myelin sheath formation.Conventional mechanisms that regulate these pathways include the integrated unfold protein response and endoplasmic reticulumassociated degradation, which are often dysregulated in neurodegenerative disease (Wu et al., 2020).Similarly, the role of macroautophagy in myelin sheath turnover has recently been shown to be essential to prevent neuronal and oligodendrocyte cell death(Aber et al.2022).As the loss of mature myelinating oligodendrocytes in the CNS likely contributes to a number of neurological diseases, understanding the pathomechanisms of oligodendrocyte dysfunction and how it can be overcome for remyelination is a high priority.Whole fields of study toward oligodendrocyte remyelination are highlighted in multiple sclerosis literature, a canonical demyelinating neurodegenerative disease.Excitingly, through many high-throughput chemical screening approaches,numerous small molecules have been identified that stimulate the formation of mature myelinating oligodendrocytes from OPCs (Lariosa-Willingham et al., 2016) that can be explored in future studies of oligodendrocyte maturation resilience in neurodegenerative disease models.
Because of advances in RNA sequencing resolution,oligodendrocyte signatures in neurodegenerative disease contexts were identified primarily through either single-cell or single-nucleus RNA sequencing(scRNAseq and snRNAseq, respectively).Per NIH guidelines, there are numerous datasets available for deep interrogation of common pathomechanisms.By meta-analysis of this omic data, one could generate a valuable resource of oligodendrocyte and neuronal changes within neurodegenerative contexts.This would provide an opportunity for indepth analysis of shared signatures, which could illuminate biomarker and therapeutic prospects for neurodegenerative diseases.While an incredibly powerful tool, it is important to keep in mind with RNAseq that differential gene expression does not necessarily translate to protein expression, which in turn does not confer function.Functional assays,such as transmission electron microscopy orin vivotwo/three-photon imaging to evaluate myelination or cellular interactions (Figure 1bandd), are necessary to be able to assess the significance of changes in RNA and protein expression.As previously mentioned,oligodendrocytes have many functions in the CNS beyond their canonical role as myelinating cells.These are often overlooked or downplayed when implicating oligodendrocytes in disease.Recent studies utilizing genetic reporter mouse lines andin vivotwo-photon microscopy (Figure 1aandb) have shown that myelin remodeling plays a role in learning (Yalcin and Monje, 2021).These findings could have important implications for memory, as well as dementia, a common symptom in many neurodegenerative diseases.The underlying mechanisms and regulation of this remodeling are not yet clear, therefore the effect of oligodendrocyte dysfunction on this noncanonical role of myelin remains elusive.When investigating circuit-level, cellular, and molecular mechanisms, it is also imperative to keep in mind the heterogeneity of oligodendrocytes, regarding maturation state, local environment, function, and transcriptome (Seeker and Williams, 2022).Through combining various levels of analysis to look at the big picture, it will be possible to begin to untangle the causal or consequential effects of disease-associated oligodendrocytes in neurodegenerative diseases.
Oligodendrocyte function can be grossly disrupted in disease, and just as importantly, so too can their interactions with other cell types.Oligodendrocytes,and OPCs in particular, have been shown to interact quite extensively with other glial cells, as well as with neurons (Yalcin and Monje, 2021).Microglia and astrocytes are involved in myelin maintenance and remodeling, however, the signaling pathways that drive these interactions remain incomplete.OPCs act as more than just a progenitor pool to replace mature oligodendrocytes, they form gap junctions with astrocytes and synapse onto neurons, yet little is known about the significance of these connections.An even larger gap in this knowledge is how diseaseassociated oligodendrocyte signatures affect these interactions and if disruption contributes to disease pathogenesis.Signaling pathways and systems-level circuitry involved in these intercellular connections have been relatively unexplored in disease, and the impact of oligodendrocyte dysfunction on either has been even less so.Oligodendrocytes are known to be sensitive to the microenvironment they reside in(Sherafat et al., 2021), providing a heterogeneity of sorts to the cell population.This is vital to keep in mind, as oligodendrocyte behaviorin vitromay not be representative ofin vivophenotypes when local environments and cellular interactions are considered.In summary, there are still many unanswered q u e st i o n s re ga rd i n g d i s e a s e-a s s o c i ate d oligodendrocyte signatures and their contributions to pathogenesis.How do all aspects of oligodendrocyte heterogeneity (microenvironment, maturation state, function, and transcriptome) contribute to systems, cellular, and molecular mechanisms of normal oligodendrocyte function and intercellular interactions? How might these mechanisms be dysfunctional in disease, and what are the significance and impact of these differences? To establish a broad,yet in-depth understanding of oligodendrocytes,we need to be able to perturb these cells at stages throughout their maturation.Disease phenotypes are a natural place to start investigating how disease pathology disturbs the endogenous function and activity of oligodendrocytes.However, investing time and resources into creating tools to selectively manipulate aspects of oligodendrocyte mechanisms and local interactions, such as conditional genetic mouse strains or oligodendrocyte-specific optogenetic tools and viral delivery systems, will be necessary to push this field forward.These types of methods will help define the necessary and/or sufficient behaviors of oligodendrocytes that will lead to unprecedented understanding of neuron-glia interactions that shape the form and function of the central nervous system.This work was supported in part by National Institutes of Health R01-NS122751 and U01-NS106670 to HSM.
Kristen H.Schuster,
Hayley S.McLoughlin*Department of Neurology, University of Michigan,Ann Arbor, MI, USA
*Correspondence to:Hayley S.McLoughlin, PhD,hayleymc@med.umich.edu.https://orcid.org/0000-0003-4279-2758(Hayley S.McLoughlin)https://orcid.org/0000-0002-8896-3623(Kristen H.Schuster)
Date of submission:October 28, 2022
Date of decision:December 20, 2022
Date of acceptance:January 7, 2023
Date of web publication:January 30, 2023
https://doi.org/10.4103/1673-5374.368302
How to cite this article:Schuster KH,McLoughlin HS (2023) Disease-associated oligodendrocyte signatures in neurodegenerative disease: the known and unknown.Neural Regen Res 18(10):2192-2193.
Open access statement:This is an open access journal, and articles are distributed under the terms of the Creative Commons AttributionNonCommercial-ShareAlike 4.0 License,which allows others to remix, tweak, and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.

- 中国神经再生研究(英文版)的其它文章
- From static to dynamic: live observation of the support system after ischemic stroke by two photon-excited fluorescence laser-scanning microscopy
- MicroRNAs in mouse and rat models of experimental epilepsy and potential therapeutic targets
- The generation and properties of human cortical organoids as a disease model for malformations of cortical development
- Nanotechnology-based gene therapy as a credible tool in the treatment of Alzheimer’s disease
- Detection of Alzheimer’s disease onset using MRI and PET neuroimaging: longitudinal data analysis and machine learning
- A pancreatic player in dementia: pathological role for islet amyloid polypeptide accumulation in the brain