
Pediatric GNAO1encephalopathies:from molecular etiology of the disease to drug discovery
2023-03-23 08:13VladimirKatanaevJanaValnohovaDenisSilachevYonikaLarasatiAlexeyKoval
中国神经再生研究(英文版) 2023年10期
Vladimir L.Katanaev, Jana Valnohova, Denis N.Silachev, Yonika A.Larasati,Alexey Koval
Gαo is the major G protein in neurons, where it transduces signals from numerous G proteincoupled receptors (GPCRs) such as D2 dopamine,μ-opioid, M2 muscarinic, or α2-adrenergic receptors.In 2013, the first mutations inGNAO1, the gene encoding Gαo, were described in pediatric patients with encephalopathies(Nakamura et al., 2013), suffering from movement disorders, epileptic seizures, and developmental delay.As of today, over 200 patients have been identified asGNAO1mutation carriers (https://gnao1.org/) mainly thanks to the availability of the whole exome sequencing technique.The mutations are typically single amino acid substitutions that mostly occur de novo, however unique cases of inheritance of the mutations are reported as well.In the OMIM (Online Mendelian Inheritance in Man) catalog, the two following disorders are associated with the heterozygous mutation inGNAO1(the homozygous mutations have never been detected in humans; thus, it is likely that mutations in both alleles are lethal):
The developmental and epileptic encephalopathy 17 (DEE17, OMIM 615473), also called Early infantile epileptic encephalopathy (EIEE17),manifests with intractable (not easily controlled by medicine) seizures, which start within the first weeks after birth.Some patients have burstsuppression patterns of seizures and high-voltage abnormal brain activity alternating with periods of very little activity, described as Ohtahara syndrome.Many patients also suffer from developmental delay.
The other disorder is neurodevelopmental disorder with involuntary movements (NEDIM,OMIM 617493), a disease characterized by the early onset of hyperkinetic involuntary movements and delayed psychomotor development.The symptoms can be very severe, often preventing the patients to be able to sit, walk, speak or eat.Since patients of both DEE17 and NEDIM are affected in their movement abilities even though it sometimes manifests just as a low muscle tone(hypotonia),GNAO1encephalopathy can be viewed as the disease that involves movement disorders with (DEE17) or without (NEDIM)seizures.A comprehensive recent review on the clinical features of GNAO1 encephalopathy can be found in (Axeen et al., 2021).
In neurons, wild-type Gαo is present in two locations: the plasma membrane and the Golgi apparatus (Solis et al., 2017, 2022).We hypothesized that at the cellular level, the manner how these two localizations are affected by the specific mutations determines the difference in the clinical manifestations.For example, mutations that decrease the ability of the protein to reach the plasma membrane result in the DEE17 disease that includes seizures (Solis and Katanaev,2018), while those still localizing properly to both intracellular destinations yet aberrant in activity –to NEDIM with movement disorders dysfunctions and developmental delays without detectable seizures.Indeed, the Glu246Lys mutation leading to the movement disorder without seizures is found to localize normally (Larasati et al., 2022),while the Gln52Arg mutation reveals aberrant localization with a decrease from the plasma membrane, causing both movement disorders and seizures (Solis et al., 2021).
While a more systematic analysis of the intracellular localization ofGNAO1encephalopathy variants is under way, we further questioned what is the molecular outcome of the pathologic Gαo mutations.Being a G protein, Gαo cycles through the activation-deactivation phases regulated by guanine nucleotides: loading with guanosine triphosphate (GTP) (stimulated by GPCRs) renders the protein active, while GTP hydrolysis through the intrinsic GTPase activity (that can further be stimulated by GTPase-activating proteins)returns it to the basal, guanosine diphosphatebound and inactive state.We focused on the three most frequent point mutations found in the encephalopathy patients – Gly203Arg, Arg209Cys,and Glu246Lys – and studied their GTP binding and hydrolysis properties, in comparison with the wildtype Gαo.Noteworthy, these mutations either directly fall into the catalytic Switch II region of Gαo (Gly203Arg, Arg209Cys), or affect this region(Glu246Lys) via a salt bridge (Larasati et al., 2022).Resultingly, these mutations reveal an almost complete loss of the GTPase capacity.Modeling and molecular dynamics simulation permitted to identify the likely mechanism of this loss of activity as the aberrant positioning of the catalytic loop and specifically of the key for hydrolysis residue,Gln205.The expectation could thus be that these pathologic mutants are constitutively active akin to the classical (not found in patients) Gln205Leu variant that is unable to interact with βγ-subunits and to be activated by GPCRs, yet persistently interacts with signaling partners such as RGS19.
Surprisingly, we found that the three pathologic mutant proteins behave in a nearly opposite to the Gln205Leu way in all respects except for the inability to hydrolyze GTP.First, their GTP binding rate is increased as compared to wildtype (or Gln205Leu) 10- to 100-fold, in the order Glu246Lys→Arg209Cys→Gly203Arg.Secondly,the interaction with βγ-subunits does not drop to zero as it is for the constitutively active variant:staying the same for Arg209Cys, it is slightly decreased for Gly203Arg, but is paradoxically increased for the Glu246Lys mutant.Thirdly,the same multidirectional change in the binding efficiency is observed for another binding partner of Gαo, AGS3.And lastly, the binding capacity of the three pathologic mutants towards RGS19 is essentially lost.Interestingly, the GTP binding rate has a clear correlation with the mutants’behavior towards these interaction partners: βγ and AGS3 binding progressively increases in the order of Gly203Arg→Arg209Cys→Glu246Lys,while the interaction with RGS19 decreases in the same order.We may thus conclude that for these switch II-affecting mutations, it is the GTP uptake rate (but not the rate of hydrolysis),which emerges as a predictor of the degree of aberration of the signaling activity (Larasati et al.,2022).To complement this analysis, we applied bioluminescence resonance energy transfer to study the ability of the pathologic mutants to interact with and respond to activation by neuronal GPCRs.Interestingly, the three mutants showed divergent deficiencies in this regard, with responsiveness to e.g.D2 dopamine, μ-opioid,and M2 muscarinic receptors decreasing in the order Glu246Lys→Arg209Cys→Gly203Arg (Larasatiet al., 2022), expanding the earlier finding on the reduced coupling of the pathogenic mutants to the D2 dopamine receptor (Muntean et al.,2021).Cumulatively, our findings identify the pathologic Gαo mutants as predominantly GTPloaded, yet unable to adopt the properly activated conformation and thus transmit the signal.
These analyses shed light on the molecular etiology of the disease.In order to advance the understanding of how these deficiencies translate into neurological disease, it is desired to create experimental animal models that would reproduce the pathological course of the disease in humans.We established such models in two systems: the fruit fly Drosophila melanogaster and the mouse.Drosophila is a popular model in neuroscience due to the existence of well-developed methods of genetic manipulation, the complex behaviors of the fruit fly, and well-described neuroanatomy.As the first step towards modeling of the disease in Drosophila, we performed humanization of the Gαo locus in the fruit fly (Savitsky et al., 2020).Remarkably, the human protein was found to fully replicate the endogenous Gαo functions, as the humanized flies were viable and fertile, depicted normal development, motor activity, longevity,and learning capacities (Savitsky et al., 2020),while Gαo loss-of-function in Drosophila is early embryonic lethal (Katanaev et al., 2005).The success in Gαo humanization is a rare achievement in Drosophila genetics and validated this organism to create a model of GNAO1 encephalopathy.To establish aGNAO1encephalopathy model in the fruit fly, we applied the CRISPR-Cas9 mutagenesis to introduce the pathogenic Gly203Arg mutation into the Drosophila Gαo.The heterozygousGly203Argflies manifested significant motor dysfunction, decreased life span, and certain brain degeneration (Larasati et al., 2022),which is reminiscent of motor dysfunction and morphological changes in the brain in human patients (Schirinzi et al., 2019); in the homozygous setting, the mutation was lethal at the second larval stage.
In parallel, we used CRISPR-Cas9 mutagenesis to establish mouse models based on the Gly203Arg and Cys215Tyr mutations, giving severe vs.mild clinical manifestations in human patients(Silachev et al., 2022).Unfortunately, theGly203Arg/+ genotype proved to be neonatal lethal in mice.Instead, theCys215Tyr/+ mice were viable but revealed strong hyperactivity and hyperlocomotion in a panel of behavioral assays, without signs of epilepsy, recapitulating the patients’manifestations and similar to theArg209His/+ mouse model (Larrivee et al., 2020).Subsequent analysis of the morphology of E18.5 embryos of theCys215Tyr/+ andGly203Arg/+mutant genotypes revealed common features: a decrease in the number of neural progenitor cells,as well as impaired migration and differentiation of projection neurons.These findings permitted us to conclude thatGNAO1encephalopathy is to a large extent a neurodevelopmental disorder.Our data expand the understanding of the critical role of Gαo in the development of the nervous system and of the disease etiology.
Current treatments forGNAO1encephalopathy patients attempt to at best partially alleviate symptoms, but do not go to correcting the core of the disease (Axeen et al., 2021).We wondered whether our insights into the molecular mechanisms of the disease could bring us to design a screening platform and to launch a drug discovery campaign forGNAO1encephalopathy.The platform we built screened for molecules able to restore the GTP hydrolysis activity of mutant Gαo, using a Food and Drug Administrationapproved drug library of some 2700 compounds(Larasati et al., 2022).Remarkably, this screening resulted in the identification of zinc pyrithione as a compound restoring the GTPase activity.Zinc pyrithione is used for topical dermatological applications and cannot be applied systemically.However, we further delineated the active part of zinc pyrithione as the Zn2+ion.Replacing Mg2+in the active center of Gαo, it brings the displaced Gln205 back to the vicinity of the γ-phosphate of GTP in the three pathologic mutants, restoring GTP hydrolysis and cellular interactions, with a negligible effect on the wild-type Gαo (Larasati et al., 2022).Being approved as a dietary zinc salt supplementation for a number of neurological conditions, zinc appears as a safe and promising therapy to be offered to theGNAO1patients with one of the three mutations we studied:Glu246Lys, Arg209Cys, and Gly203Arg.At the same time, some other mutations inGNAO1proved to be irresponsive to zinc (our unpublished observations).Importantly, dietary zinc supplementation rescued the motor function and longevity of theGly203Argmutant flies.
Overall, our study set a landmark in developing and implementing a novel workflow towards rare genetic diseases.This workflow (Figure 1) starts with the patient and combines a set of molecular,biochemical, and cellular investigations to obtain insights into the molecular etiology of the disease as caused by a specific mutation.Another important outcome of these investigations is the establishment of a high-throughput screening platform, in which Food and Drug Administrationapproved drugs are screened to identify the candidates restoring the deficient functioning of the protein affected by the specific mutation.The hit compound is then validated in thein vitroandin vivomodels of the disease.The lucky outcome of this workflow is the identification of an approved drug that can be repositioned for the new indication – the rare genetic disease we started from, thus closing the loop of this bedto-bench-to-bed, patient-personalized research(Figure 1).We are currently in the process of applying this workflow to other mutations inGNAO1encephalopathy (that are zinc-insensitive)and also to other neurological disorders.
This work was supported by the grant number 21-15-00138 from the Russian Science Foundation to VLK and DNS.
Vladimir L.Katanaev*, Jana Valnohova,Denis N.Silachev, Yonika A.Larasati,Alexey Koval*Translational Research Centre in Oncohaematology,Department of Cell Physiology and Metabolism,Faculty of Medicine, University of Geneva, Geneva,Switzerland (Katanaev VL, Valnohova J, Silachev DN, Larasati YA, Koval A)Institute of Life Sciences and Biomedicine, Far Eastern Federal University, Vladivostok, Russia(Katanaev VL, Silachev DN)Department of Functional Biochemistry of Biopolymers, A.N.Belozersky Research Institute of Physico-Chemical Biology, Moscow State University, Moscow, Russia (Silachev DN)
*Correspondence to:Vladimir L.Katanaev, PhD,Vladimir.Katanaev@unige.ch; Alexey Koval, PhD,Alexey.Koval@unige.ch.https://orcid.org/0000-0002-7909-5617(Vladimir L.Katanaev)https://orcid.org/0000-0002-8920-4426(Alexey Koval)
Date of submission:November 21, 2022
Date of decision:January 5, 2023
Date of acceptance:January 18, 2023
Date of web publication:March 3, 2023
https://doi.org/10.4103/1673-5374.369106 How to cite this article:Katanaev VL, Valnohova J,Silachev DN, Larasati YA, Koval A (2023) Pediatric GNAO1 encephalopathies: from molecular etiology of the disease to drug discovery.Neural Regen Res 18(10):2188-2189.
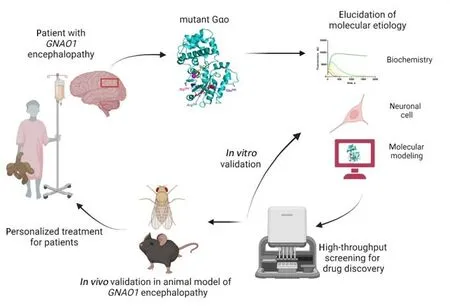
Figure 1| Workflow of the bed-to-bench-to-bed, patient-personalized research in rare genetic diseases.Created with BioRender.com.
Open access statement:This is an openaccess journal, and articles are distributed under the terms of the Creative Commons AttributionNonCommercial-ShareAlike 4.0 License,which allows others to remix, tweak, and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.
Open peer reviewer:Thomas Wirth, Département de Neurologie, Hôpital de Hautepierre, Hôpitaux Universitaires de Strasbourg, France.
Additional file:Open peer review report 1.

- 中国神经再生研究(英文版)的其它文章
- From static to dynamic: live observation of the support system after ischemic stroke by two photon-excited fluorescence laser-scanning microscopy
- MicroRNAs in mouse and rat models of experimental epilepsy and potential therapeutic targets
- The generation and properties of human cortical organoids as a disease model for malformations of cortical development
- Nanotechnology-based gene therapy as a credible tool in the treatment of Alzheimer’s disease
- Detection of Alzheimer’s disease onset using MRI and PET neuroimaging: longitudinal data analysis and machine learning
- A pancreatic player in dementia: pathological role for islet amyloid polypeptide accumulation in the brain