
Oxidative stress in retinal pigment epithelium degeneration: from pathogenesis to therapeutic targets in dry age-related macular degeneration
2023-03-23 08:13MeenakshiMauryaKiranBoraAlexandraBlomfieldMadelinePavlovichShuoHuangChiHsiuLiuJingChen
中国神经再生研究(英文版) 2023年10期
Meenakshi Maurya, Kiran Bora, Alexandra K.Blomfield, Madeline C.Pavlovich, Shuo Huang, Chi-Hsiu Liu, Jing Chen
AbstractAge-related macular degeneration is a primary cause of blindness in the older adult population.Past decades of research in the pathophysiology of the disease have resulted in breakthroughs in the form of anti-vascular endothelial growth factor therapies against neovascular age-related macular degeneration; however, effective treatment is not yet available for geographical atrophy in dry agerelated macular degeneration or for preventing the progression from early or mid to the late stage of age-related macular degeneration.Both clinical and experimental investigations involving human agerelated macular degeneration retinas and animal models point towards the atrophic alterations in retinal pigment epithelium as a key feature in age-related macular degeneration progression.Retinal pigment epithelium cells are primarily responsible for cellular-structural maintenance and nutrition supply to keep photoreceptors healthy and functional.The retinal pigment epithelium constantly endures a highly oxidative environment that is balanced with a cascade of antioxidant enzyme systems regulated by nuclear factor erythroid-2-related factor 2 as a main redox sensing transcription factor.Aging and accumulated oxidative stress triggers retinal pigment epithelium dysfunction and eventually death.Exposure to both environmental and genetic factors aggravates oxidative stress damage in aging retinal pigment epithelium and accelerates retinal pigment epithelium degeneration in age-related macular degeneration pathophysiology.The present review summarizes the role of oxidative stress in retinal pigment epithelium degeneration, with potential impacts from both genetic and environmental factors in age-related macular degeneration development and progression.Potential strategies to counter retinal pigment epithelium damage and protect the retinal pigment epithelium through enhancing its antioxidant capacity are also discussed, focusing on existing antioxidant nutritional supplementation, and exploring nuclear factor erythroid-2-related factor 2 and its regulators including REV-ERBα as therapeutic targets to protect against age-related macular degeneration development and progression.
Key Words:age-related macular degeneration; antioxidant; nuclear factor erythroid-2-related factor 2;oxidative stress; retinal pigment epithelium; REV-ERBα
Introduction
Advances in modern healthcare have significantly increased the average life span of humans in the last two centuries.Although ultimately beneficial, this advanced longevity also brings the diseases associated with aging into the limelight, such as cardiovascular diseases, cancer, dementia, and others.In the eye, aging is associated with visual impairment caused by cataracts and also retinal diseases such as age-related macular degeneration (AMD) (Klein et al., 1992; Vingerling et al., 1995; Friedman et al., 2004).As a major cause of blindness in the elderly population (> 65 years old), AMD affects the macula,the central area of the retina responsible for fine color vision.Worldwide, the number of AMD patients is estimated to be ~200 million in 2020, and with the aging population, the number is expected to increase to 288 million by 2040 (Wong et al., 2014).
Development of AMD occurs progressively over decades and based on the symptoms AMD can be classified into three stages: early, intermediate, and late or advanced (Figure 1AandB).Early and intermediate stages show increasing size of drusen, which are lipid-enriched subretinal deposits, and pigmentary changes in retinal pigment epithelium (RPE), a monolayer of phagocytic and nourishing cells beneath the retina.The yellow subretinal deposits known as drusen were first described in 1854 from the German word“druse”meaning“geode”, yet its connection with AMD was not fully recognized until the 20thcentury (de Jong, 2016).Now considered a key characteristic feature of AMD, drusen are formed due to gradual extracellular accumulation of lipid, protein, and/or cellular debris between RPE and Bruch’s membrane in an age-dependent manner (Dithmar et al., 2001; Hageman et al., 2001).Currently, the size of drusen serves as a classification marker of the clinical progression or worsening of AMD (Ferris et al., 2013).In addition,there are two types of AMD, dry (atrophic) and wet (neovascular).In dry AMD,which accounts for 80–90% of patients, the late advanced stage is marked by the presence of geographic atrophy (GA), characterized by localized sharply delineated atrophic area of RPE and choroidal vessels.On the other hand,wet AMD, a less common neovascular form of late AMD accounting for about 10–15% of AMD patients, features classic choroidal neovascularization (CNV)with associated subretinal fluid and hemorrhages (Table 1).Both GA and CNV are considered key characteristics of the late or advanced stage of AMD for dry and wet forms respectively.While GA in dry AMD results in gradual loss of vision, CNV in wet AMD frequently results in acute and profound vision loss in elderly patients (Bressler, 2002).
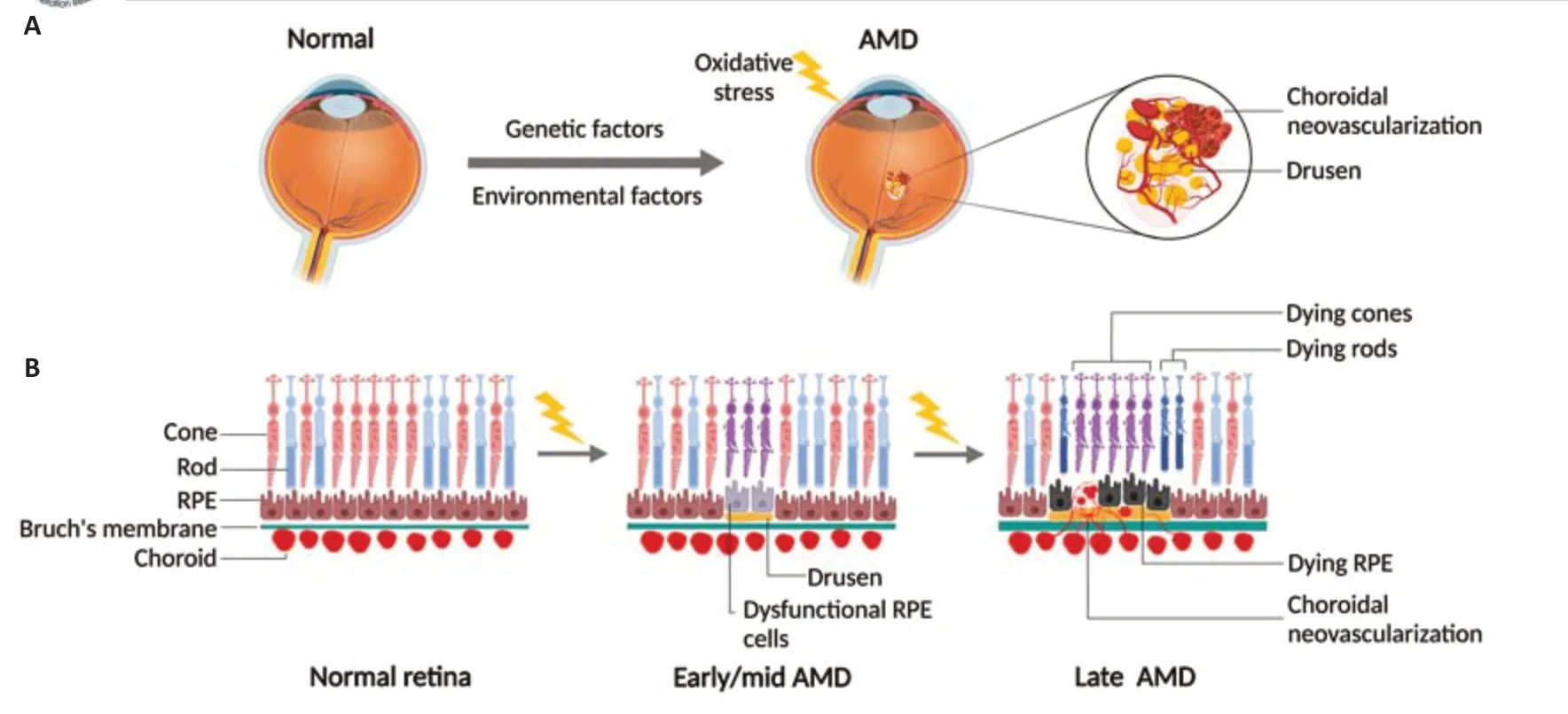
Figure 1|Schematic representation of pathological and cellular changes in RPE-retina complex with AMD development and progression.
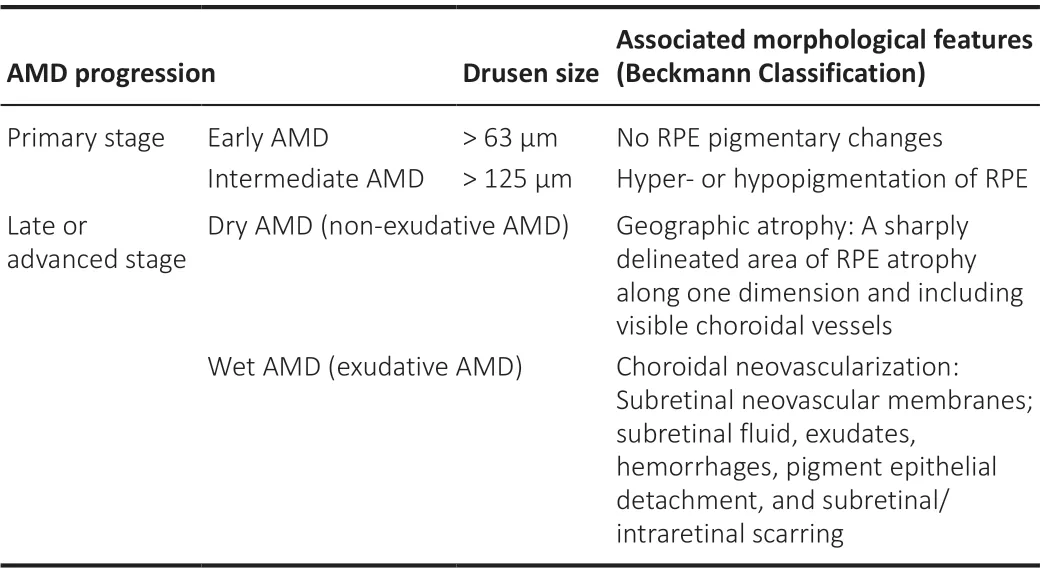
Table 1 |Stages of AMD progression and classification
Currently, there is no cure for AMD nor successful ways to prevent its onset.Available treatment options for neovascular AMD include photodynamic therapy, laser photocoagulation, and anti-vascular endothelial growth factor(VEGF) therapies that have been successful in the past two decades, although some patients remain unresponsive to treatment.For the more prevalent dry AMD, ongoing trials include anti-complement, stem cell and RPE replacement therapies; however, few treatments are available to slow down the disease progression, with the only evidence-based means being dietary intake of antioxidant nutrients.The use of antioxidant vitamins is endorsed by promising findings from two Age-Related Eye Disease Study (AREDS) trials,which exhibited their protective effects on reducing the risk of progression to advanced AMD by ~25% over 5 years (AREDS, 2001; AREDS2, 2013).These trial results highlight the important role of countering oxidative stress and damage in dry AMD pathogenesis and disease prevention, although the AREDS formula is not universally recommended by a few other studies with no effects or unanticipated health risks (Evans, 2008; Datta et al., 2017),which potentially reflects the interaction of genetics and vitamin supplement(Vavvas et al., 2018).Apical junctions between RPE cells also form the outer blood-retinal barrier,which selectively transports nutrients and metabolic waste between the neural retina and the choroid, through Bruch’s membrane on the basal side of RPE (Strauss, 2005; Fields et al., 2020).In addition, RPE secretes VEGF towards the choroid to maintain the health of choroidal vessels (Saint-Geniez et al.,2009).RPE also protects the neural retina from light-induced damage through light absorption by melanin and lipofuscin.All these biochemical processes require active oxidative metabolism, hence, making RPE highly susceptible to oxidative damage.AMD is a multifactorial disease with many factors in addition to aging contributing to the disease progression, including environmental agents and genetic predisposition, involving oxidative stress, chronic inflammation,innate immunity (complement pathway), and lipid metabolisms (Jun et al., 2019; Fleckenstein et al., 2021; Zhang and Wong, 2021).The complex interaction among these mechanisms is still under intense investigation to fully elucidate the disease pathogenesis, with the possibility that different pathological processes may take place simultaneously to result in distinct pathologies in multiple AMD-associated cell types (Hadziahmetovic and Malek, 2020; Bird, 2021).This review summarizes the current understanding of AMD pathogenesis with a specific focus on the role of oxidative stress in RPE degeneration in dry AMD.Relevant advances from both clinical and experimental studies are discussed to highlight potential new strategies to counter oxidative damage and protect RPE.
Search Strategy and Selection Criteria
The PubMed database was used to search and select relevant literature, using the following combinations of keywords: age related macular degeneration,antioxidants, oxidative stress, retinal pigment epithelium, Age-Related Eye Disease Study, nuclear factor erythroid-2-related factor 2, and REV-ERBα.
Oxidative Damage Is a Major Cause of Retinal Pigment Epithelium Degeneration in Dry Age-Related Macular Degeneration
RPE plays a vital role in maintaining retinal health
RPE, a monolayer of pigmented epithelial cells between the retina and choroid, has a vital function in supporting photoreceptors and vision with multiple trophic tasks.On the apical side, they recycle photoreceptor outer segments via phagocytosis and assist the visual cycle through retinoid cycling.
RPE cells are exposed to high levels of reactive oxygen species
Generation of reactive oxygen species (ROS) is an integral part of normal cellular metabolism and physiology, in order to maintain cellular energy demands and function.ROS generation in turn serves as the framework for redox signaling to control its balance.RPE is exposed to high levels of oxidative stress, primarily from excess mitochondrial ROS, and phagocytosisrelated oxidative events (Datta et al., 2017; Brown et al., 2018).
The choroid vasculature carries high O2pressure passing across RPE to the retina, to meet its metabolic demand.RPE contains a large number of mitochondria, which create a highly oxidative environment, with the production of ROS including superoxide, hydrogen peroxide, and hydroxyl radical (Zorov et al., 2014; Brown et al., 2019).In addition to the major ROSproducing site in mitochondrial complexes I & III, complexes II and IV of the electron transport chain system also generate ROS, which leaks from the mitochondrial membrane in normal physiological conditions (Liu et al., 2002).Such ROS leakage tends to increase with aging and can exacerbate the agedependent ROS accumulation (Cadenas and Davies, 2000; Liang and Godley,2003; Sohal and Orr, 2012).
Lipid oxidation from phagocytosed outer segments (enriched with polyunsaturated fatty acids), and photo-oxidation due to constant light stimulation, contribute to excess ROS in RPE additionally (Cai et al., 2000;Lambros and Plafker, 2016; Brown et al., 2018).Photoreceptor outer segment disk engulfment is primarily carried out by RPE through a lysosome-efficient system that leads to the accumulation of lipofuscin debris, a marker of cellular senescence, within the lysosome with aging.Environmental and lifestyle factors such as smoking and an unbalanced western diet, serve as additional sources of chemical and dietary oxidants, contributing to high ROS levels in RPE (Malek et al., 2012; Brown et al., 2018; Figure 2).
RPE redox homeostasis is maintained by an antioxidant self-defense system
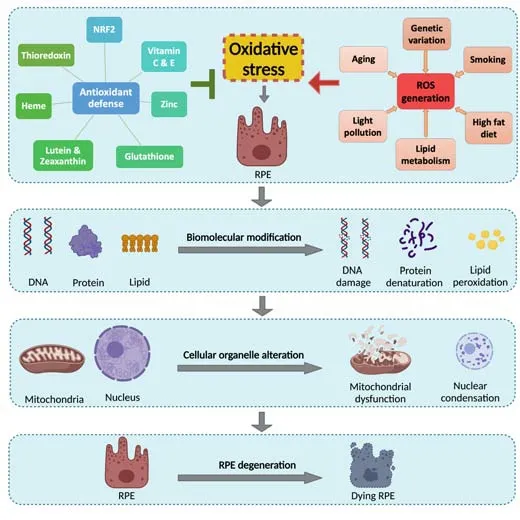
Figure 2|Schematic figure illustrating the redox balance between antioxidant pathway and ROS-inducing factors in the generation of oxidative stress causing RPE cell degeneration.
To counter the ROS, RPE has well-synchronized signaling mechanisms including antioxidants such as glutathione and heme, antioxidant vitamins,and redox proteins including thioredoxin.Moreover, the elimination of ROS in RPE is carried out primarily by a cascade of nuclear factor erythroid-2-related factor 2 (NRF2)-dependent antioxidant self-defense enzymes, including superoxide dismutase (SOD), catalase, and peroxidase, in addition to free radical scavengers (Cai et al., 2000).This system arms RPE with the ability to quench superoxide and peroxide moieties, along with other free radicals, to maintain cellular homeostasis.During aging this defense mechanism may be impaired or overwhelmed, leading to excess ROS buildup and hence oxidative damage (Suh et al., 2004; Reisman et al., 2009; Gounder et al., 2012; Volonte et al., 2013; Zhang et al., 2015a; Figure 2).In addition to antioxidants, autophagy may also contribute to the redox defense in RPE cells.In AMD, RPE specifically shows increased ROS levels,susceptibility to lipid peroxide-induced protein modification, and subsequent oxidative stress (Beatty et al., 2000; Kaemmerer et al., 2007).Compared to the normal young retina, autophagosomes, autophagy flux, and autophagyrelated proteins such as light chain 3 (LC3)-II/LC3-I ratio levels increase with aging.Gradual accrual of toxic proteins, damaged organelles, and drusen deposits leading to RPE cell dysfunction and death, owing to impaired lysosomal digestion or autophagy has been linked with AMD pathogenesis (Wang et al., 2009; Mitter et al., 2014).In fact, LC3-II/LC3-I ratio fails to increase in late-stage AMD retina and RPE (Golestaneh et al.,2017).Considering the role of autophagy in lipofuscin clearance, diminished autophagic activity might also lead to increased ROS, enhanced lipofuscin accumulation, and subsequent RPE dysfunction and death, hence increasing the risk of GA in AMD.
Oxidative stress might also downregulate RNase DICER1, deficiency of which was suggested to lead to Alu RNA toxicity, NLRP3 inflammasome activation,and RPE atrophy in AMD (Kaneko et al., 2011; Tarallo et al., 2012; Kaarniranta et al., 2020).Given the essential role of DICER1 in microRNA biogenesis, the potential role of miRNA dysregulation in RPE redox control and degeneration is also emerging (Intartaglia et al., 2020; Liu et al., 2020; Urbańska et al., 2022).Several microRNAs show potential in the modulation of RPE homeostasis, function, and autophagy and may become dysregulated in AMD RPE, such as miR-184, miR-29, and miR-1273g (Murad et al., 2014; Cai et al.,2019; Hyttinen et al., 2021) or by their inhibition such as miR-302d-3p (Jiang et al., 2018).Since the present review focuses on the oxidative stress in RPE,readers may refer to recent reviews (Jun et al., 2019; Intartaglia et al., 2020;Hyttinen et al., 2021; Du and Palczewski, 2022; Urbańska et al., 2022) for details on the interplay of microRNAs in oxidative stress and AMD.
Oxidative damage contributes to RPE degeneration
Although reactive oxygen intermediates are mainly the byproducts of physiological processes and part of a normal feedback loop in redox signaling,overexposure of ROS stimuli and impaired antioxidative defense due to aging can result in ROS accumulation.Subsequent oxidative stress and excess free radicals result in oxidative damage to macromolecules such as DNA, proteins,and lipids.In particular, lipid oxidation can alter the normal function of cellular membrane lipid and lipoprotein, and trigger inflammation.Together, these macromolecular oxidative damages can lead to the breakdown of intracellular organelles like mitochondria, and lysosomes, consequently resulting in the death of RPE and photoreceptor cells (Cai et al., 2000; Lambros and Plafker,2016; Brown et al., 2019; Figure 2).
RPE aging and degeneration lead to vision loss in AMD
Cellular aging manifests as both structural deterioration and compromised function.In RPE, signs of aging can be seen as an alteration in microvilli and basal folding structure, loss of melanin pigment, and accumulation of cellular debris and lipofuscin.These abnormalities in RPE near the macular area that activate the slow RPE dysfunction and degeneration cascade are often considered part of the triggers for AMD development (Bird, 2021).
Since one of the main functions of RPE is the circadian engulfment of the spent rod outer segment (Young and Bok, 1969; LaVail, 1976), any disturbance in this vital recycling process, either due to RPE dysfunction or alienated phagocytosis turnover with aging, may trigger ROS-derived lipid and protein accumulation and cross-link.These molecular changes can also lead to structural and functional changes in mitochondria in RPE.Compared to normal aging RPE, AMD RPE reveals reduced mitochondrial density, reduced adenosine 5’-triphosphate production, and increased loss of cristae with mitochondrial DNA damage (Kaarniranta et al., 2019).In fact, global proteome analysis of RPE from AMD patients confirms the alteration in a vast number of mitochondrial system proteins (Nordgaard et al., 2006), strengthening the idea of RPE mitochondrial dysregulation and damage-driven AMD pathogenesis (Fisher and Ferrington, 2018; Tong et al., 2022).
Bruch’s membrane adjacent to RPE also takes part in this process as agedependent degeneration in collagen and elastin impairs extracellular matrix maintenance, thereby lowering the rate of extracellular wastage clearance(Spraul et al., 1999).Together with RPE dysfunction, lipid-laden drusen forms,resulting in thickening of Bruch’s membrane (Curcio, 2018; Bird, 2021),which further hinders metabolic exchange between RPE and choroid and may aggravate deposition.Drusen thickening and deposition also trigger chronic inflammation and immune system activation that further worsen the architectural integrity of the RPE-retina complex (Gehrs et al., 2006; Datta et al., 2017), resulting in the eventual loss of trophic support by RPE for adjacent photoreceptors leading to their degeneration or death (Strauss, 2005), and subsequent gradual loss of central vision in AMD (Haddad et al., 2006; Rattner and Nathans, 2006; Figure 1B).
Studies on Environmental and Genetic Risk Factors Support the Pathogenic Role of Oxidative Damage in Age-Related Macular Degeneration
The pathogenic role of oxidative stress is reflected by environmental and lifestyle risk factors of AMD
Numerous environmental factors contribute to accelerated aging and AMD development.The association of sunlight exposure with AMD development was first highlighted by the Beaver Dam Eye Study (Cruickshanks et al., 1993),suggesting the potential risk of photo-oxidation.Apart from aging, smoking acts as a major non-genetic contributing factor in macular atrophy.Smoking is significantly associated with high retinal pigment and exudative macular degeneration in both females (odds ratio [OR]: 2.50) and males (OR: 3.29)active smokers with respect to non-smoker or ex-smoker patients (Klein et al.,1993).
The idea of oxidative factors functioning as an etiological component in AMD development is supported by gene-environmental interaction studies,such as smoking (Smith and Hansch, 2000; Rangasamy et al., 2004).Clinical investigation has suggested that smoking, paired with aging, results in an increased risk of neovascular AMD and vision loss in patients (Chew et al.,2014a).The clinical outcomes were also supported by experimental studies.RPE function requires an immense adenosine 5’-triphosphate supply fulfilled by a large number of mitochondria, with the price of constant exposure in a highly oxidative environment.Chemicals in cigarette smoke cause decreased activity of mitochondrial subunits, and increased mitochondrial superoxide production, resulting in mitochondrial dysfunction (Strunnikova et al., 2004;Jia et al., 2007; Bertram et al., 2009).Chronic exposure to cigarette smoke also compromises the in-built redox machinery within the eye to weaken redox balance.Increased lipid peroxidation (4-HNE) further amplifies oxidative stress and accelerates RPE degeneration (Klein et al., 1998; Cano et al., 2010;Ni Dhubhghaill et al., 2010).
Besides smoking, early AMD development is also accelerated by heavy alcohol consumption and hypertension, all of which are associated with increased oxidative stress and antioxidant imbalance in clinical trials (Seddon et al., 2001; Kuan et al., 2021; Zhang et al., 2021) and are detrimental to RPE.The influence of dietary patterns on AMD was also considered as food is the primary source of dietary vitamins and other antioxidants.The association of early AMD prevalence decreases with plant and seafood-based diet (OR:0.7) and increases with dairy and refined grain-rich (OR: 1.56), respectively(Chiu et al., 2014).The relative risk (RR) of progression of AMD increases with high-fat consumption, irrespective of source, whether it is plant-based (RR:2.29) or animal-based (RR: 3.82) high-fat diet (Seddon et al., 2003).The level of lipid saturation also correlates with the likelihood of AMD progression; for saturated fat (RR: 2.09), monounsaturated fat (RR: 2.21), polyunsaturated fat(RR: 2.28), and trans unsaturated fat (RR: 2.39), respectively (Seddon et al.,2003).In fact, smoking (Rahman and MacNee, 1996; Smith and Hansch, 2000;Rangasamy et al., 2004) and high-fat diet intake are considered preventable AMD risk factors (Mares-Perlman et al., 1995; Seddon et al., 2001), and both are linked with increased oxidative stress, supporting the detrimental role of oxidative stress in AMD pathogenesis.
Genetic risk factors of AMD suggest only susceptibility but not causality
Even though aging is the strongest AMD-associated risk factor, many epidemiological studies have highlighted the shared impact of genetics,ethnicity, and family history.The theory of a genetic component of AMD(De Jong et al., 1997; Seddon et al., 1997; Klaver et al., 1998) was supported by studies of monozygotic twins with significantly increased concordance rate compared to dizygotic twins (Dosso and Bovet, 1992; Klein et al., 1994;Meyers, 1994; Hammond et al., 2002; Grizzard et al., 2003).Additionally,AMD prevalence has been associated with ethnicity, i.e., higher in Caucasian populations compared to non-Caucasians (Klein et al., 2006; Bressler et al., 2008).Family history may also play a role, with an OR: 2.4 to 19.8-fold increase in prevalence in individuals with a family history of AMD as compared to those without (Seddon et al., 1997; Smith and Mitchell, 1998),hinting a genetic link.
Advances in molecular genetics provided much-needed evidence since the impact of a genetic component in innate immunity (complement system)on AMD prevalence was discovered and investigated intensely (Zhang and Wong, 2021).Several proteins of the complement system have been identified in drusen found in aging and AMD eye (McHarg et al., 2015).Genes involved in innate immunity, and particularly complement regulation, such as complement factor H (CFH) (Edwards et al., 2005; Haines et al., 2005; Klein et al., 2005), complement 3 (C3), complement 2/complement factor B (Gold et al., 2006; Francis et al., 2009), and complement factor H-related 1, 3 (Spencer et al., 2008) exhibit significant positive association in AMD development and progression.It is estimated that CFH variants, with decreased CFH function,may contribute to approximately 50% of the risk of developing AMD, with much from a missense mutation (Y420H) (Edwards et al., 2005).
In addition to complement, single nucleotide polymorphisms in two closely located, major AMD-associated genes were identified at chromosome 10q26: high-temperature requirement factor A1, serine peptidase 1 (HTRA1)(rs11200638 G→A polymorphism), and age-related maculopathy susceptibility 2 (ARMS2) (rs10490924 G→T polymorphism).Both show strong linkage disequilibrium and increase the risk of AMD by about the same magnitude(Tong et al., 2010), making it difficult to differentiate them.The importance of HTRA1 is supported by its cellular localization within the RPE and drusen in the AMD eyes (Yang et al., 2006).Increased expression of HTRA1, a serine protease, may potentially compromise the structural integrity of Bruch’s membrane, and promote RPE atrophy, whereas ARMS2, a mitochondrial protein, has an unknown function (Kanda et al., 2010).Additional genes with different cellular functions are also associated with AMD risk.For instance,genes related to lipid metabolism pathways such as hepatic lipase, and apolipoprotein E (Klaver et al., 1998; Souied et al., 1998; Baird et al., 2004;Zareparsi et al., 2004; Neale et al., 2010), in immune response, including CD36 and Toll-like receptor-3 and -4 (Zareparsi et al., 2005; Yang et al., 2008),and in retinal function adenosine 5′-triphosphate-binding cassette transporter 4 (ABCA4) (Fritsche et al., 2012; Wu et al., 2015), were also associated with AMD risk.
Despite the strong genetic link, these identified AMD-associated genes only demonstrate susceptibility without proven causality, in contrast to monogenic diseases where a single mutation is sufficient.Given the multi-factorial and complex nature of AMD and its late-in-life onset, one may speculate that genetic risk factors may interact with environmental/lifestyle factors that may precipitate the cellular damage under oxidative stress which accumulates with age, resulting in the risk of AMD development and progression.
Interaction of genetic-oxidative stress response may suggest different levels of genetic susceptibility to oxidative damage
Even with the tremendous effort in investigating AMD genetics, genetic variations alone do not accurately predict AMD progression, as highlighted by monozygotic twin studies, where smoking and dietary nutrient intake dictates disease progression along aging (Seddon et al., 2011).This suggests a significant contribution of environmental factors-induced oxidative stress which may function as modifiers, and also the possibility of geneenvironmental interaction determining in part the genetic susceptibilities.Oxidative stress may invoke innate immunity in the eye, through the binding of CFH with malondialdehyde, a common lipid peroxidation product in drusen (Weismann et al., 2011; Alic et al., 2020).The AMD-associated CFH variant H402 strongly reduces its binding affinity to malondialdehyde,suggesting a mechanistic link (Weismann et al., 2011).Similarly, the strong interaction of genetics, including CFH and ARMS2, with antioxidant nutritional supplementation (Vavvas et al., 2018), further suggests a genetics-oxidative stress link that may modify AMD risk.
Additional AMD-associated genes encode proteins comprising the redox scavenging machinery in the eye, such as mitochondria, the main source of energy and ROS generation in RPE.Polymorphism in mitochondrial DNA of MTND2*LHON4917G (4917G) and complex-1 subunit is remarkably associated with AMD development (Canter et al., 2008; SanGiovanni et al.,2009), as well as mitochondrial antioxidant enzymes (Mrowicka et al., 2017),suggesting that compromised antioxidant system prevails the path for RPE degeneration.Additional studies uncovering the link between genetic and environmental factors pertaining to oxidative stress response will provide further insights on AMD pathogenesis.
Enhancing Antioxidant Capacity in Retinal Pigment Epithelium Is a Promising Strategy to Protect against Age-Related Macular Degeneration Progression
Dietary intake of antioxidant vitamins for AMD is endorsed by AREDS &AREDS2 studies
In 1988, a small randomized clinical trial reported for the first time that oral supplementation with zinc, a trace element and essential micro-nutrient,can reduce AMD progression (Newsome et al., 1988).Given the role of zinc in antioxidant response, that trial motivated the use of antioxidant supplementation to protect against AMD development or progression.Later,the beneficial role of antioxidant vitamins in AMD was supported by two large clinical trials: Age-Related Eye Disease Study (AREDS) & Age-Related Eye Disease Study 2 (AREDS2).
Given the chronic nature of AMD, to evaluate whether long-term administration of antioxidants and other dietary supplements may exert greater visual protection, National Eye Institute sponsored AREDS with 3640 individuals for an average of 6.3 years between 1992 and 2001 in the USA,with two primary objectives: first, to investigate the natural course of AMD and age-related cataracts; second, to assess the effects of antioxidants i.e.,vitamins C, E, beta-carotene, and zinc on AMD.Original AREDS formula consists of vitamin C (500 mg), vitamin E (400 IU), beta-carotene (15 mg),zinc as zinc oxide (80 mg), and copper as cupric oxide (2 mg) (AREDS, 2001).AREDS showed that nutritional supplementation with antioxidants (vitamins C, E, and beta-carotene) and zinc are protective and reduce the risk of progression to late AMD by 25% in 5 years, by scavenging ROS to ameliorate oxidative stress (AREDS, 2001).In a follow-up study, the beneficial effects of a long-term (10 years) AREDS formulation diet (5 years after the trial ended)persisted in successfully delaying the progression of advanced neovascular AMD in patients with AMD in one eye, but not for the central GA (Chew et al.,2013).AREDS2 study further improved the formula with lutein (10 mg) and zeaxanthin (2 mg), both carotenoids, replacing beta-carotene, to eliminate the concern of beta-carotene in increased lung cancer risk (Chew et al.,2012; AREDS2, 2013).The AREDS2 formula showed incremental protection in intermediate-stage AMD patients compared with the original formula, further confirming the effects of antioxidant vitamins (AREDS2, 2013).Currently, the AREDS2 formula is the only scientifically proven intervention that can delay the progression of intermediate- or late-stage AMD, despite it being noneffective for people with no AMD or early AMD.However, another study has reported varying patient responses (SanGiovanni et al., 2007), potentially reflecting intricate interaction between patient genetics and nutrient intake that warrants further investigation.
Consideration of genetics-nutrient interaction is needed
Since AREDS, the possible influence of the genotype over the AREDS formula treatment response was explored by numerous studies, with findings both supporting and rejecting the idea.Early studies by Awh et al.(2013, 2015)have supported the pharmacogenomics-based AREDS supplement selection in AMD patients.They suggested that the use of zinc and antioxidant should be separated based on specific risk or non-risk allele (CFH/ARMS-2) in patients, with zinc only for those with non-CFH risk, 1–2 risk allele for ARMS-2, and in contrast antioxidant only for those with no-risk ARMS-2 and 1–2 risk allele for CFH (Awh et al., 2013; Awh et al., 2015).These claims were refuted by a separate analysis of AREDS participants by Chew et al., suggesting that AREDS supplements exert similar levels of protection against AMD risk among all genotype groups without any specific influence by CFH risk allele (Chew et al., 2014b), rejecting the need of genetic testing-based ARDES formulation assigning altogether (Chew et al., 2015).
Other later studies found a positive association between AREDS supplementderived AMD protection and allele variations.For instance, in neovascular AMD patients, AREDS supplements resulted in a lower risk of AMD progression to an advanced stage in high-risk ARMS2 (TT) and non-risk CFH allele (TT) group, but not in CFH high-risk allele (CC) group, yet similar results were not seen in GA (Seddon et al., 2016; Vavvas et al., 2018).On the contrary, AREDS supplement accelerated the rate of AMD progression in highrisk CFH genotype (Vavvas et al., 2018); however, these results were subjected to debate as many other studies, both from AREDS reports and independent analysis of AREDS cohorts, have rejected the genotype-dependent association between AMD progression and AREDS formulation (Assel et al., 2018; van Asten et al., 2019).
Given the debate, more studies will be needed to be able to draw definitive conclusions.Yet consideration of the genetics-nutrient interaction is still warranted, particularly considering the use of supplements in different races and ethnic groups with diverse genetic backgrounds.The occurrence of late AMD is significantly higher in the white population compared to other races in North America (Schachat et al., 1995; Leske et al., 2004; Varma et al.,2004; Restrepo et al., 2014), and they were the primary participants in both US-based AREDS trials.In view of the different dietary intake and genetic makeup in other countries, data related to the nutritional effects on other ethnic populations is still much lacking.This outcome, while await future studies, would have significance on the impact of genetic-nutrient interaction.Nevertheless, current insights obtained have greatly promoted interest in experimental and translational investigations on targeting oxidative stress as a potential therapeutic strategy to protect against AMD in animal models.
Experimental Studies on Oxidative Stress and Cellular Defense: Nuclear Factor Erythroid-2-Related Factor 2 as a Key Antioxidant Enzyme Regulator
NRF2 is a central transcriptional regulator of antioxidant enzymes
Transcriptional regulation of many redox pathway proteins is moderated by NRF2 (encoded by thenfe2l2gene), a master transcriptional regulator of key antioxidant enzymes.In physiological conditions, NRF2 is sequestered by Kelch-like ECH-associated protein 1 (KEAP1) and negatively regulated through ubiquitination and proteasomal degradation (McMahon et al., 2003).Excess ROS triggers a conformational modification in KEAP1, resulting in the release and subsequent nuclear translocation of NRF2, which then binds to MAF protein and an antioxidant response element on the promoter site of target genes to induce their transcription.These include direct antioxidant enzymes,such as SOD1&2 and catalase, which directly neutralize superoxide and hydrogen peroxide (H2O2), as well as phase 2 enzymes regulating glutathione and thioredoxin levels, and xenobiotic enzymes, e.g.glutathione peroxidase,heme oxygenase 1&2, and NADPH quinine oxidoreductase 1 (Figure 3).Through regulating these antioxidant defense enzymes, NRF2 mediates the redox balance in many cell types including RPE, which has been extensively studied in preclinical animal models of AMD.
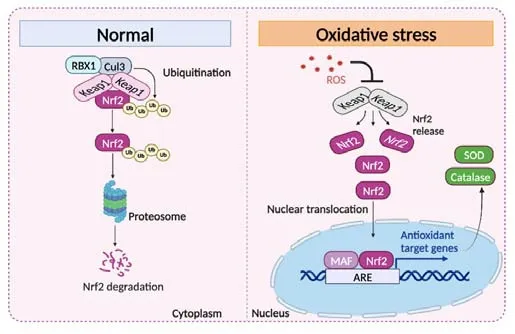
Figure 3|A cartoon illustration of NRF2 signaling.
Experimental studies on AMD highlight the role of NRF2 in RPE oxidative defense
While there are no perfect animal models of AMD that reproduce all pathological features in humans, experimental studies have benefited much from animal models showing key features of AMD-like pathologies.These include inflammation (Cfh–/–and/orCx3cr1–/–mice) (Coffey et al.,2007; Combadiere et al., 2007), impaired lipid function (transgenicApoE4with high-fat diet, and macrophage-specificABCA1andABCG1deficiency)(Ramkumar et al., 2010; Ban et al., 2018), and oxidative stress and defense(Sod1–/–,Nrf2–/–,AhR–/–, or carboxyethylpyrrole immunization) (Imamura et al., 2006; Hollyfield et al., 2008; Ramkumar et al., 2010; Zhao et al., 2011;Hu et al., 2013), each displaying some features of AMD (Ramkumar et al.,2010; Pennesi et al., 2012).For example,Sod1–/–mice (Imamura et al., 2006),lacking Cu, Zn-superoxide dismutase-1 (Sod1), a critical enzyme involved in removing superoxide radicals, and hence important for oxidative stress response, exhibited increased drusen with age (from 6–18 months), thickened Bruch’s membrane, disrupted RPE integrity and choroidal neovascularization at 10–12 months of age, with reduced ERG waves, and progressive retinal cell death (Hashizume et al., 2008).In addition, systemic SOD2 knockdown mice also exhibited RPE degeneration and photoreceptor disorganization (Kasahara et al., 2005; Justilien et al., 2007), and RPE-specific deletion of SOD2 led to RPE and photoreceptor metabolic alteration, dysregulation, and dysfunction(Brown et al., 2019).Moreover, sodium-iodate-induced RPE oxidative damage is commonly used as an acute animal model to investigate mechanisms of oxidative damage in RPE (Franco et al., 2009).Together these animal models highlight the role of antioxidant enzymes and oxidative damage in the development of RPE degeneration and AMD-like features.
Given the central role of NRF2 in antioxidant enzyme regulation, a potential role of NRF2 in RPE degeneration is widely investigated.In aging RPE,induction of NRF2 signaling is compromised with oxidative stress (Sachdeva et al., 2014).Nrf2–/–mice developed AMD-like ocular pathologies including sub-RPE deposits with RPE degeneration (Zhao et al., 2011) and these degenerative events accelerated after mild white light exposure (Wang et al., 2019) and increased cigarette smoke-induced RPE damage (Cano et al., 2010), suggesting a key role of NRF2 in RPE oxidative damage and potentially AMD development.In addition,Nrf2–/–mice also developed agerelated cataracts (Rowan et al., 2021), highlighting the importance of NRF2-dependent antioxidant signaling in aging eyes.Recently, NRF2 deficiency was also found to impact the mitochondrial antioxidant response by influencing the pentose phosphate pathway in RPE (Cano et al., 2021).Global double knockout of NRF-2 and PGC-1α (peroxisome proliferator-activated receptor gamma coactivator-1 alpha) in mice resulted in significant age-dependent RPE degeneration with mitochondrial damage as well as increased expression of inflammatory and autophagy markers mimicking dry-AMD pathologies(Felszeghy et al., 2019).In addition, a low glycemic diet protectedNrf2–/–mice against AMD-related pathologies, via enhanced antiglycation and anti-oxidative machinery (Rowan et al., 2020).These studies established a pivotal role of NRF2 in RPE and AMD pathophysiology (Cai et al., 2021), yet transcription of NRF2 itself can be subjected to regulation by other factors involved in redox sensing and protection.
REV-ERBα: a newly discovered transcriptional regulator of NRF2 and antioxidant defense in RPE
One such example of NRF2 regulators is REV-ERBα, a redox-sensitive nuclear transcription factor primarily involved in circadian rhythm regulation and metabolism.REV-ERB is a family of redox-sensing nuclear receptors functioning as ligand (heme)-sensitive transcriptional repressors (Raghuram et al., 2007; Kojetin and Burris, 2014; Brown et al., 2018) and a critical sensor of the tissue redox environment in modulating cellular oxidative capacity(Pardee et al., 2009; Gupta and Ragsdale, 2011; Ragsdale et al., 2012; Woldt et al., 2013; Carter and Ragsdale, 2014).A dynamic thiol-disulfide mechanism regulates their ligand binding affinity in a redox-sensitive (thiol-disulfide exchange) manner (Pardee et al., 2009; Ragsdale et al., 2012; Woldt et al.,2013; Carter and Ragsdale, 2014).REV-ERB at a reduced (dithiol) state binds ligand with much higher affinity (5-fold) than the oxidized disulfide state(Gupta and Ragsdale, 2011); therefore, oxidative stress acts through this thioldisulfide redox switch to lead to oxidation of cysteine(s) residues in REV-ERB,thus causing ligand dissociation to dampen the receptor’s transcriptional activity.
REV-ERBα (also known as Nr1d1) is an essential regulator of many biological processes including circadian rhythm, lipid and glucose metabolism,macrophage inflammatory response, and mitochondrial biosynthesis (Cho et al., 2012; Solt et al., 2012; Lam et al., 2013; Woldt et al., 2013; Zhang et al.,2015b), some of which are highly relevant to RPE health and AMD.Ligand binding to REV-ERBα determines its interaction with other transcriptional cofactors (NCoR and HDAC3) (Zhang et al., 2015b).Upon binding to the specific DNA response element (RORE) in the regulatory region of the target genes,these co-factors together mediate the expression of target genes, particularly circadian clock genes (Woldt et al., 2013; Zhang et al., 2015b).However, for metabolic target gene expression, it was suggested that REV-ERBα recruits corepressors, such as HDAC3, which tethers REV-ERBα with other tissue lineagespecific transcription factors to mediate the expression of specific metabolic genes tailoring to the need of specific tissues (Woldt et al., 2013; Butler and Burris, 2015; Zhang et al., 2015b).
In the eye, shRNA-targeted knockdown ofRev-erbαin developing mouse eye with sub-retinal shNR1D1 injection led to pan-retinal spotting with decreased visual function (Mollema et al., 2011).REV-ERBα regulates retinal gene transcription in conjunction with NR2E3 (Mollema et al., 2011), and also retinal visual processing and light sensitivity (Ait-Hmyed Hakkari et al., 2016),as well as RPE circadian genes (Milicevic et al., 2019).Genetic overexpression of REV-ERBα as a modifier gene rescuesNr2e3-associated retinal degeneration inrd7mice (Cruz et al., 2014).Rev-erbα–/–mice have increased numbers of intrinsically photosensitive retinal ganglion cells and demonstrate increased light sensitivity, suggesting a role of REV-ERBα in retinal visual processing (Ait-Hmyed Hakkari et al., 2016).
Recently, we found that REV-ERBα is a direct transcriptional regulator of NRF2 (Huang et al., 2022).Both systemic and RPE cell-specific deficiencies of REV-ERBα result in the downregulation of NRF2 and its target antioxidant enzymes, SOD1 and catalase.More importantly, REV-ERBα-deficient mice exhibit age-dependent ocular pathologies including fundus lesions, thickened Bruch’s membrane, sub-retinal drusenoid deposits, RPE degeneration, and photoreceptor disorganization, suggesting thatRev-erbα–/–mice represent a new animal model of dry AMD.In an oxidative stress-exposed (NaIO3)model of RPE damage, REV-ERBα-deficient mice are more susceptible to RPE oxidative damage, whereas treatment with REV-ERBα agonist, SR0009 protects against RPE damage with upregulation of NRF2, SOD1, and catalase(Huang et al., 2022).These findings together suggest a protective role of REV-ERBα against RPE degeneration in response to oxidative stress and that targeting REV-ERBα may help protect RPE in dry AMD (Figure 4).In other studies, REV-ERBα agonists were found to be cytoprotective in general, and effective in promoting skeleton muscle cell survival, suppressing diabetesinduced metabolic changes, and atherosclerosis (Solt et al., 2012; Woldt et al., 2013; Sitaula et al., 2015; Ait-Hmyed Hakkari et al., 2016) and promoting the targeted killing of cancer cells (Sulli et al., 2018).REV-ERBα agonists may thus serve as attractive drug target candidates for developing therapeutics for not only AMD but also other diseases.

Figure 4|A graphical illustration of REV-ERBα-dependent transcriptional regulation of antioxidant response through NRF2 in RPE relevant to AMD pathophysiology.
Promoting NRF2 to counter oxidative damage in RPE
Because of its essential role in sensing redox stress, NRF2 has become a molecular target for eye disease protection and particularly RPE protection against oxidative stress.Indeed, RPE-specific NRF-2 overexpression via adeno-associated virus vector delivery preserves RPE morphology and cone photoreceptors in mouse models of retinitis pigmentosa (Wu et al.,2021).In addition, intravitreal delivery of adeno-associated virus containing an NRF2-derived peptide protected RPE and partially restored visual function in the NaIO3-induced RPE oxidative injury model by inducing antioxidant pathway and dampening inflammation (Ildefonso et al., 2016).Moreover, overexpression of NRF2 also protects retinal neurons including photoreceptors and retinal ganglion cells in rodent models of photoreceptor degeneration and nerve crush, more than viral delivery of SOD2 and catalase(Xiong et al., 2015), suggesting the promise of targeting NRF2 in protecting retinal neurons and RPE against oxidative damage.
Many studies explored both upstream and downstream redox regulatory mechanisms of NRF2 in RPE using variousin vitroandin vivomodel systems.These models rely on the induction of oxidative stress by free radical generating substances, such as sodium iodate-induced oxidative stress model (Han et al., 2020; Tang et al., 2021), white/blue light exposure (Liu et al., 2021; Xie et al., 2021), cigarette smoke extract exposure (Huang et al.,2015) and H2O2-induced oxidative damage model (Du et al., 2020; Ma et al., 2021; You et al., 2021).Together these work in cell culture (Huang et al.,2015; Du et al., 2020; Liu et al., 2021; Ma et al., 2021; Tang et al., 2021; Xie et al., 2021; You et al., 2021) and animal-based models (Han et al., 2020; Xie et al., 2021) discover additional regulators of NRF2 signaling, just like REVERBα.For instance, X box-binding protein 1, an endoplasmic reticulum stressinducible transcription factor (Huang et al., 2015; Chen et al., 2018) and microphthalmia-associated transcription factor (Han et al., 2020) also act as a novel upstream modulator of NRF2 against oxidative stress damage in RPE.Targeting microRNA is also emerging as an effective strategy to modulate upstream NRF2 signaling cascade to counter oxidative stress in RPE, for instance, targeting cullin 3 by miR-601 against H2O2-induced stress (Chen et al., 2019), or Keap1 by microRNA-141 (Cheng et al., 2017) and miR-125b (Liu et al., 2022).
Other additional studies focused on the effects of pharmacological modulators with chemical compounds to influence NRF2 signaling in RPE preservation.Smoking, in particular, impairs NRF2 signaling in aging RPE cells (Suzuki et al., 2008; Wang et al., 2014).Exposure of cigarette smoke extract to NRF2-deficient RPE cells exhibited elevated oxidative stress due to decreased expression of IDH2 and pentose phosphate pathway genes (Cano et al., 2021).In the blue light-driven oxidative stress model of RPE cell culture and BALB/c mice, treatment with lipoxin A4, an antioxidant lipid, reduces the blue light-driven oxidative stress by upregulating heme oxygenase-1 (HO-1) and nuclear translocation of NRF-2 (Xie et al., 2021).In the H2O2-induced oxidative damage model, RPE cells were pre-treated with different chemical compounds: including ferrerol, a rhododendron-derived NRF2 activator(Ma et al., 2021), catalpol, an iridoid glucoside fromRehmannia glutinosaLibosch plant root dry extract (You et al., 2021), and phillyrin, extracted from Chinese herbaceous plantForsythia suspense(Thunb.)Vahfruits with anti-inflammatory and antioxidant properties (Du et al., 2020).Together these compounds revealed NRF2 activating potential and subsequent elevation of its downstream antioxidant enzymes: catalase, HO-1, NADPH dehydrogenase,SOD and increased glutathione, and hence inhibition of endogenous and exogenous apoptotic pathways.In these studies, inhibition of Keap1/NRF2 protein complex formation, nuclear translocation of NRF2, and elevation in HO-1 expression levels were consistently shown among many oxidative stress models, suggesting that HO-1 upregulation by NRF2 is one of the key hierarchical axis against the oxidative stress in RPE (Shivarudrappa and Ponesakki, 2020; Chen et al., 2021; Ma et al., 2021; Tang et al., 2021; You et al., 2021; Zhao et al., 2021), in addition to other NRF2-mediated antioxidant enzymes.Together these studies demonstrated the translational potential of promoting activation and expression of NRF2, its upstream regulators,and downstream target enzymes, to counter oxidative stress-induced RPE damage.
Summary
As dry AMD can progress to neovascular AMD, treating patients at the early stage of dry AMD could potentially prevent or slow down the disease progression to the late devastating stages of GA and CNV.Protection of RPE against oxidative damage is vital for slowing AMD progression and preservation of vision.In recent years, stem cell transplantation and RPE patch transplantation have shown great promise in patients with AMD (Mandai et al., 2017; Yang et al., 2022), yet safety and efficacy will await more studies.Preventive measures to halt or slow down AMD progression, however, are still very negligible.On that note, long-term dietary intake of antioxidant supplements suggested by AREDS & AREDS2 studies significantly reduces the progression to late-stage AMD, highlighting the importance of oxidative damage in AMD pathogenesis.However, there are no current pharmacological approaches to slow or stop dry AMD progression or curb oxidative injury in RPE.
With both human genetics and experimental studies strongly supporting the role of redox imbalance in RPE oxidative injury during aging, limiting oxidative stress may thus act as a preventive and therapeutic measure against RPE oxidative damage and degeneration.Key transcriptional regulatory factors of antioxidant pathways such as NRF2 can be targeted to drive the cascade of synchronous expression of various antioxidant enzymes and pro-survival proteins to protect RPE in AMD.Identification of novel upstream regulators of NRF2, such as REV-ERBα, adds new ways to modulate antioxidant pathways and enhance RPE antioxidant self-defense system.Future work can be directed towards the revelation of the intertwined interaction of immune and inflammatory, metabolic, and antioxidant pathways in AMD.That will further improve our understanding of AMD pathogenesis and allow the development of new effective therapeutic strategies to counter RPE oxidative damage for disease management.
Author contributions:MM and JC conceived and wrote the review; MM and CHL prepared the table and figures; all authors critically reviewed, edited and approved the manuscript.
Conflicts of interest:The authors declare no conflicts of interest.No conflicts of interest exist between Mass Lions Eye Research Fund Inc.and publication of the manuscript.
Data availability statement:The data are available from the corresponding author on reasonable request.
Open access statement:This is an open access journal, and articles are distributed under the terms of the Creative Commons AttributionNonCommercial-ShareAlike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.
Open peer reviewer:Lawrence J.Rizzolo, Yale University School of Medicine,USA.
Additional file:Open peer review report 1.

- 中国神经再生研究(英文版)的其它文章
- From static to dynamic: live observation of the support system after ischemic stroke by two photon-excited fluorescence laser-scanning microscopy
- MicroRNAs in mouse and rat models of experimental epilepsy and potential therapeutic targets
- The generation and properties of human cortical organoids as a disease model for malformations of cortical development
- Nanotechnology-based gene therapy as a credible tool in the treatment of Alzheimer’s disease
- Detection of Alzheimer’s disease onset using MRI and PET neuroimaging: longitudinal data analysis and machine learning
- A pancreatic player in dementia: pathological role for islet amyloid polypeptide accumulation in the brain