
Mitochondrial dysfunction as a target in spinal cord injury: intimate correlation between pathological processes and therapeutic approaches
2023-03-23 08:13JulietaSchmidtctorRamiroQuint
中国神经再生研究(英文版) 2023年10期
Julieta Schmidt, Héctor Ramiro Quintá
AbstractTraumatic spinal cord injuries interrupt the connection of all axonal projections with their neuronal targets below and above the lesion site.This interruption results in either temporary or permanent alterations in the locomotor, sensory, and autonomic functions.Damage in the spinal tissue prevents the re-growth of severed axons across the lesion and their reconnection with neuronal targets.Therefore, the absence of spontaneous repair leads to sustained impairment in voluntary control of movement below the injury.For decades, axonal regeneration and reconnection have been considered the opitome of spinal cord injury repair with the goal being the repair of the damaged long motor and sensory tracts in a complex process that involves: (1) resealing injured axons; (2)reconstructing the cytoskeletal structure inside axons; (3) re-establishing healthy growth cones;and (4) assembling axonal cargos.These biological processes require an efficient production of adenosine triphosphate, which is affected by mitochondrial dysfunction after spinal cord injury.From a pathological standpoint, during the secondary stage of spinal cord injury, mitochondrial homeostasis is disrupted, mainly in the distal segments of severed axons.This result in a reduction of adenosine triphosphate levels and subsequent inactivation of adenosine triphosphate-dependent ion pumps required for the regulation of ion concentrations and reuptake of neurotransmitters,such as glutamate.The consequences are calcium overload, reactive oxygen species formation, and excitotoxicity.These events are intimately related to the activation of necrotic and apoptotic cell death programs, and further exacerbate the secondary stage of the injury, being a hallmark of spinal cord injury.This is why restoring mitochondrial function during the early stage of secondary injury could represent a potentially effective therapeutic intervention to overcome the motor and sensory failure produced by spinal cord injury.This review discusses the most recent evidence linking mitochondrial dysfunction with axonal regeneration failure in the context of spinal cord injury.It also covers the future of mitochondria-targeted therapeutical approaches, such as antioxidant molecules, removing mitochondrial anchor proteins, and increasing energetic metabolism through creatine treatment.These approaches are intended to enhance functional recovery by promoting axonal regenerationreconnection after spinal cord injury.
Key Words:adenosine triphosphate; axonal regeneration; creatine; mitochondria dysfunction;mitochondria; spinal cord injury
Introduction
Spinal cord injury (SCI) is one of the main causes of disability among young adults worldwide, with an estimated 250,000 to 500,000 new injuries each year (Khorasanizadeh et al., 2019; Liang et al., 2022).Although physiological impairments can be affected by a whole range of biological functions, the loss of motor function particularly influences the degree of self-sufficiency that affected individuals can achieve in their daily lives, directly impacting their quality of life.At the neuronal level, SCI is a traumatic pathology that damages the axonal tracts innervating spinal neurons and/or in spinal circuits, temporarily or permanently altering motor, sensory, or autonomic function (Yezierski, 2009).The axonal damage at the lesion site caused by the traumatic event disrupts the bi-directional information flow between the brain and spinal cord.For this reason, the therapeutic approach for SCI is challenging, taking into account the lack of an effective“gold standard”strategy to promote neuronal repair.Therefore, the recovery of locomotor activity after SCI is considered one of the main goals in the search for new therapies by scientists around the world.
Although axonal regeneration is considered the ultimate goal in SCI research,it would not be enough to promote full locomotor recovery (Raineteau and Schwab, 2001).This is due to the multiple deleterious effects produced not only by the primary trauma but also by the secondary damage focalized in the spinal cord.Most of the axonal tract degeneration post-injury is caused by secondary damage occurring at the primary lesion site as well as at remote areas.This secondary stage of trauma includes different processes such as hemorrhage, blood-brain barrier dysfunction, ischemia and reperfusion,low levels of oxygen partial pressure, inflammation, glial-scar formation, and mitochondrial dysfunction (Park et al., 2004; Burda and Sofroniew, 2014;Streijger et al., 2017; Ge et al., 2021; Slater et al., 2022).
The alteration in the biological function of mitochondria caused by spinal cord damage promotes oxidative stress and reactive oxygen species (ROS)formation.It also alters calcium homeostasis, creates a crisis in the energy requirements caused by an alteration in the adenosine triphosphate (ATP)production, and produces excitotoxicity.Taken together, these processes advance axonal dieback, neural circuit collapse, and activation of the biological program of necrosis and apoptotic cell death (Hirsch et al., 1998).
Additionally, spinal cord damage produces remote degeneration that appears upstream and/or downstream of the lesion, because these areas are functionally connected to the primary lesion site, exacerbating neurological deficits (Lotze et al., 1999; Latini et al., 2014; Bisicchia et al., 2017).
All of these deleterious effects explain why SCI is mostly an irreversible pathology.To date, the only treatment that has shown any success in promoting the recovery of motor function in humans is training-based rehabilitation therapy (Harkema et al., 2012).Nevertheless, this therapy seems to be only partially effective in patients with incomplete motor lesions(Dietz et al., 1994; Harkema, 2008).
However, in the case of severe injuries, even with a certain degree of local circuit preservation, only a modest improvement occurs, which is insufficient to trigger muscular activity.In other words, no voluntary movement is possible in the absence of supraspinal control.Therefore, voluntary motor control depends on healthy descending axons from motor cortex neurons that relay supraspinal centers, jointly contacted with the spinal circuits responsible for controlling movement, located above and below the lesion site (Quinta,2021).
To promote the preservation, repair, and reconnection of motor axons avoiding neuronal degeneration by SCI, it is essential not only to promote the re-activation of cytoskeleton dynamics, leading to activation of a reparative program but also to obtain“metabolic energy”.This is necessary to promote the polymerization of actin, tubulin, and intermediate filaments to start regenerating and sprouting in the more distal segment of the damaged axons(Jacob and McQuarrie, 1996), preventing axonal dieback (Seif et al., 2007).Furthermore, synaptic contact depends on the production of metabolic energy, a process triggered by healthy mitochondria in the axonal shaft.
Several well-established therapeutic approaches have shown contributions to promoting re-polymerization of the cytoskeleton after injury.For instance,treatment with molecules such as Galectin-1, Netrin-1, Taxol, and Epothilone B and D has exhibited exciting results in the promotion of axonal regeneration(Hellal et al., 2011; Bradke et al., 2012; Quinta et al., 2014, 2016; Ruschel et al., 2015; Sandner et al., 2018).
However, these approaches need an active metabolism to obtain the energy required to trigger the regeneration.According to recent preclinical therapeutics in this field, two types of interventions targeting the mitochondria are being tested as potential treatments for SCI (Han et al.,2020).Since neurons are metabolically active cells, the first strategy is to maintain energetic function and ATP cellular distribution in the damaged axon by a proper fusion-fission process that is necessary to maintain mitochondrial function.The fusion is likely to protect function, providing an equal distribution of metabolites as well as enabling protein complementation.In contrast, fission enhances the distribution of mitochondria along the cytoskeleton tracts and promotes the isolation of damaged segments of mitochondria (Chen and Chan, 2009).
The second strategy is the administration of a bioenergetics compound such as creatine that rapidly regenerates ATP from adenosine diphosphate,independent from mitochondrial transport, acting like an energy facilitator and improving the reparative process after SCI (Tarnopolsky and Beal, 2001).Moreover, they could act together with other molecules that stimulate the re-growth of axonal growth cones, axolemma, and axoplasm to reinforce the reparative process.The aim of this article is to discuss the most recent evidence linking mitochondrial dysfunction with axonal regeneration failure after spinal cord injury.
Search Strategy and Selection Criteria
In this narrative review, we examined mitochondrial dysfunction in SCI and potential therapeutic approaches.We used the PubMed database and Google Scholar to search for articles published between 1994 and 2022 using the following keywords or combinations: spinal cord injury, mitochondria dysfunction, ROS, ATP, BSCB in SCI, biofuel in SCI, mitochondria-targeted therapeutical approaches, creatine, axonal damage, secondary stage of injury,axons and mitochondria, axolemma/axoplasm disruption, and macrophages.
Mitochondria and the Central Nervous System
Mitochondria are intracellular energy factories in the human body.In the central nervous system (CNS), the functionality of neurons depends on mitochondrial integrity, bidirectional mitochondria movement along the axons, and mitochondria activity (Tarnopolsky and Beal, 2001; Zhou et al.,2016), since neuronal activity relies on stringent and efficient ATP production.As a source of energy, ATP maintains not only neuronal functionality but also proper propagation of membrane potential and the release and uptake of neurotransmitters in the pre-post synapsis (Scholpa and Schnellmann, 2017).Also, neurons have a limited antioxidant capacity, preventing them from maintaining proper homeostasis after damage (Adibhatla and Hatcher, 2010).Therefore, considering all of these characteristics, neuronal activity and its synaptic interactions rely heavily on a proper mitochondrial metabolism that includes mitochondria axonal distribution-transport (fission-fusion) and ATP production along the axonal shaft.However, each of these processes is altered when mitochondrial function is impaired after SCI (Slater et al., 2022).
Mitochondria in Health and in Spinal Cord Injury
At a physiological level, mitochondria are responsible for more than 90% of cellular oxygen consumption and for the production of the majority of ATP in the cell (Nicholls and Ferguson, 1992).Furthermore, mitochondria participate in the biosynthesis of fatty acids (Kastaniotis et al., 2017), cellular calcium homeostasis (Pivovarova and Andrews, 2010), and triggering apoptosis by translocation of cytochrome c from the mitochondria to the cytoplasm(Jiang and Wang, 2004; Jemmerson et al., 2005).These processes properly modulate an extensive number of cellular functions in the human body.Some examples include providing energy for proper myocardiocyte activity as well as for insulin production by pancreatic beta cells and supplying ATP that is essential for axonal growth, survival, and regeneration at the neuronal level(Nicholls and Budd, 2000).As mentioned above and limited to the topics discussed in this mini-review,mitochondria act at the neuronal level as an energy source in physiological conditions.However, in pathophysiological conditions, especially in pathologies like SCI, mitochondria dysfunction is caused by neuronal damage.Therefore, taking into account that a large amount of bio-energy is needed to restore and boost axonal regeneration in damaged neurons (Bradke et al., 2012), mitochondrial re-activation plays a key role as a power supply to support the energetically demanding regenerative process.
The dysfunction caused by axonal injury (Figure 1) decreases bioenergetic metabolism, preventing the molecular steps that lead to neuroprotection.On the other hand, the recovery of mitochondria integrity and functionality could serve as a therapeutic intervention that increases or re-establishes the energy source in the axons at the spinal cord level to overcome the damage produced by the injury (Sullivan et al., 2000; Verburg and Hollenbeck, 2008;Zhou et al., 2016; Han et al., 2020).
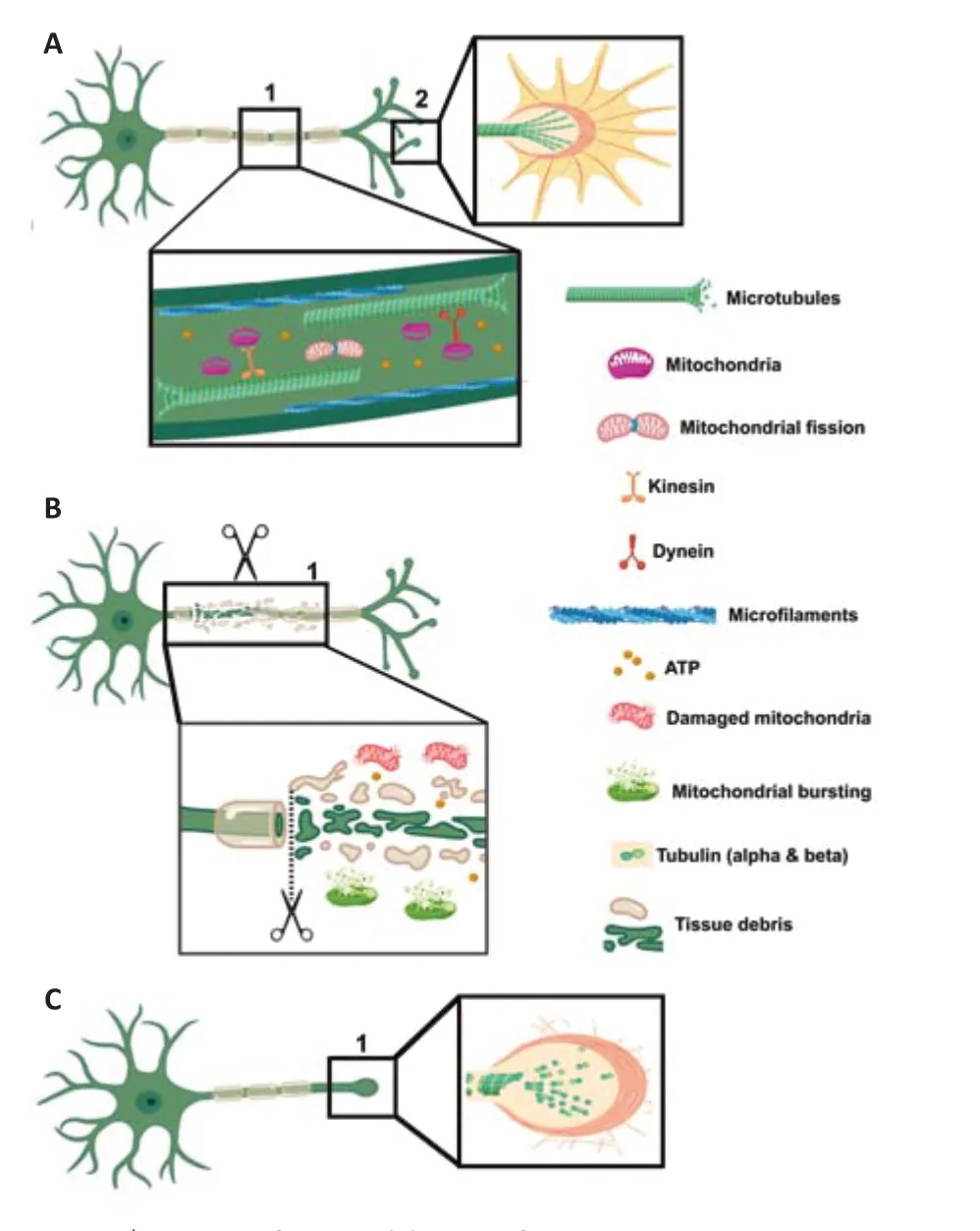
Figure 1|Cartoon of neuronal damage after SCI.
Molecular Pathways of Mitochondria Dysfunction after Spinal Cord Injury
As mentioned earlier, the primary trauma results in mechanical disruption of(1) cortical axons and propriospinal neurons (producing cellular and molecular changes at the level of cortico-spinal and rubrospinal neurons), (2) glial cells,and (3) the blood-spinal cord barrier (BSCB).Axonal rupture occurs almost instantaneously after injury, leading to the loss of some subcellular organelles(such as mitochondria) until the axonal sealing is completed in five minutes to a number of hours.The rupture of the BSCB usually takes place within five minutes and lasts up to 28 days.The whole process leads to vasoconstriction hemorrhage edema activation of the inflammatory response axonal dieback and glial cell proliferation, migration, and activation (Baptiste and Fehlings,2006; Graumann et al., 2011; Tran et al., 2018).
The degree of ischemia post-injury is proportional to the spinal cord impairments (Tator and Fehlings, 1991) that contribute directly to the negative consequences observed after BSCB rupture since it limits the paracellular and transcellular transport of different types of molecules (e.g.,insulin, albumin, and glucose).Consequently, BSCB disruption results in the infiltration of peripheral inflammatory cells and factors into the lesion site,contributing to the emergence of the secondary SCI stage (Winkler et al.,2011).The molecular cascade that triggers the secondary damage starts in the first minutes to hours after primary injury, and peaks approximately 2 weeks after SCI but could last for months.In fact, the deleterious effect produced by this secondary damage is greater than the primal insult (Tanhoffer et al.,2007; Oyinbo, 2011) because it can reach remote areas.As a consequence of secondary damage, mitochondrial dysfunction appears, contributing strongly to the progression of the pathology and preventing neuronal self-repair(Scholpa and Schnellmann, 2017; Slater et al., 2022).
Several molecular pathways are involved in the ignition and progression of the secondary stage of injury that can be divided into three distinct phases: acute, sub-acute, and chronic (Quadri et al., 2020).One of the main molecules that trigger this process is ROS, a general term given to a group of oxygen molecules derived from reduction-oxidation reactions or electronic excitation.These molecules have an intimate relationship with mitochondria,where an accumulation of ROS can be implicated in the loss of mitochondrial homeostasis, leading to energy impairment (Sies and Jones, 2020).
Very early after SCI, an increase in the mitochondrial ROS cascade is observed in the cells localized at the lesion site (Hall et al., 2016; Slater et al., 2022).The inhibition in ROS production might protect injured spinal cord tissue from mitochondrial oxidative stress, neuronal apoptosis, and axonal dieback.Similarly, it is well known that there is a direct relationship between BSCB disruption after SCI and the impairments in cell types that form the spinal cord.The evidence shows that vascular endothelial cells increase the mitochondrial ROS levels as a result of oxygen consumption after a CNS injury,which affects the integrity of BSCB since a high level of ROS impairs vascular function in regulating inflammation, cell proliferation, and mitophagy.Hence,it is necessary to maintain mitochondrial homeostasis to manage ROS production by vascular endothelial cells after SCI.
The interesting question here is how and why the endothelial cells promote ROS increases after injury.This is related to perivascular macrophages that play a key role in pathogenic neurovascular damage by increasing ROS levels (Faraco et al., 2016).After SCI, macrophage infiltration modulates neuroinflammation at the epicenter of the lesion.Although both M1 and M2 macrophage phenotypes appear at the injury site, the proinflammatory cytotoxic M1 phenotype plays a dominant role over M2 throughout the entire damage process, increasing BSCB disruption via neuroinflammation and promoting endothelial cell apoptosis in the early stages of trauma (Kigerl et al., 2009; Milich et al., 2019).From a molecular standpoint, after BSCB disruption by SCI, M1-polarized bone marrow-derived macrophages promote mitochondrial ROS formation.As Ge et al.(2021) described, this process is mediated by exosomal transfer of miR-155 followed by suppression of SOCS6-induced p65 degradation, since miR-155 is upregulated in M1-polarized macrophages to induce neuroinflammation.
Thus, there are many factors beyond mechanical trauma related to mitochondrial dysfunction after SCI that involve cellular and subcellular alteration at the neuronal level, increasing proinflammatory molecules that contribute to neuroinflammation and ROS formation.
Adenosine Triphosphate Production after Spinal Cord Injury
After spinal damage, angiogenesis is initiated as an attempt to overcome the disruption of BSCB.However, this process is not completely effective and the emerging vessels are often leaky, preventing the full delivery of nutrients into as well as the removal of waste from the injured spinal cord (Kundi et al., 2013).Additionally, the decreased oxygen supply at the epicenter of the lesion affects the ability of mitochondria to maintain homeostasis.
Mitochondrial homeostasis is closely associated with maintaining neuronal functions such as electrical homeostasis, membrane potential conduction,and the release and uptake of neurotransmitters.All of these depend on stringent and efficient ATP regulation that involves the flux of various ions across the plasma membrane (Adibhatla and Hatcher, 2010) and requires a large amount of energy.
Due to the limited intrinsic antioxidant defenses of the neuron (Adibhatla and Hatcher, 2010), its proper function relies heavily on healthy mitochondrial metabolism and ATP production, processes altered by mitochondria dysfunction in the presence of CNS damage.In the secondary damage that appears after primary SCI, the loss of mitochondrial function results in the concomitant loss of ATP along with the inactivation of ATP-dependent ion pumps that regulate ion concentration and the reuptake of the excitatory neurotransmitter glutamate.This loss of function promotes excitotoxicity and calcium overload, triggering the cell death program, a chain of events that is a hallmark of SCI (Choi and Rothman, 1990; Rowland et al., 2008; Oyinbo, 2011).The subcellular architecture of mitochondria changes after SCI, opening the mitochondria permeability transition pore that is closed under physiological conditions.This pathological process induces swelling of mitochondria,leading to the rupture of the mitochondrial outer membrane, resulting in the release of cytochrome c into the cytosol and a concomitant triggering of apoptosome formation (Miura and Tanno, 2012).Also, the alteration of mitochondria permeability transition pore permeability decreases the mitochondrial membrane potential (∆ψM), interrupting ATP synthesis (Hirsch et al., 1998).Finally, due to oxidative damage, several mitochondrial enzymes are inactivated.In particular, pyruvate dehydrogenase, a key player in the generation of acetyl-CoA, is critical for the production of NADH and FADH2(McEwen et al., 2011).Thus, spinal damage induces acute mitochondrial depolarization and ATP depletion in the damaged axons.Conversely,enhancing mitochondrial transport could prevent ATP deficit and improve regenerative capacities in the damaged neurons (Zhou et al., 2016).
Mitochondria in Axonal Damage and Repair
Axons in the spinal cord correspond to descendent and ascendant axonal tracts as well as axons from propriospinal neruons.When a traumatic event occurs in the spinal cord, like a transection, laceration, stretch, or compression, the axons suffer different types of damage (Shi and Pryor,2000; Nehrt et al., 2010; Ouyang et al., 2010; Quinta et al., 2014; Quinta and Barrantes, 2019).These different types of damage represent different degrees of disruption to the axolemma membrane up to a complete section of the axonal shaft.
In such cases, a dynamic process starts after injury that includes an attempt to rearrange the axonal plasma membrane and reactivate cytoskeleton dynamics in the axoplasm, which involves a depolymerization-polymerization phenomenon over microfilaments and microtubules (Schelski and Bradke,2017).This process encompasses a fast molecular mechanism that induces the re-sealing of the plasma membrane with the intent of promoting neuronal survival and subsequent axolemma repair.Although this repair process can occur, it is slow in the CNS, taking several minutes to several hours.By comparison, re-sealing other cell types such as fibroblasts takes seconds to minutes (Hendricks and Shi, 2014; Quinta and Barrantes, 2019).
The neuronal plasma membrane has several functions, including maintenance of intracellular homeostasis, regulation of ion exchange with the extracellular milieu, and regulation of synaptic compartments (Quinta and Barrantes,2019).Under pathological conditions like spinal cord trauma, a fast,retrograde degeneration of axons occurs.The damage in the neuronal plasma membrane produces a breach in the structure of the axolemma that alters the physiological homeostasis, increasing the permeability of ions and molecules associated with the loss of membrane integrity (Bradke et al., 2012).This promotes a sharp influx of Ca2+and other molecules to the intra-axonal space that activates proteases, modifying the normal mitochondrial function, and leading to the activation of the neuronal apoptosis program (Nguyen et al.,2005; Kilinc et al., 2009; Hendricks and Shi, 2014).
To complete the re-sealing of the axonal plasma membrane after trauma,there is a sub-membrane structure that acts synergistically by promoting the formation of the retraction bulb.This bulb corresponds to a dystrophic axonal structure characterized by presenting massive depolymerization of cytoskeleton components at the tip of the severed axon.This retraction bulb formation is a hallmark of axonal growth cone collapse (Griffin and Bradke,2020).
Axonal dieback starts from the retraction bulb, increasing the distance that the axons need to regenerate in order to re-connect with the targets localized on the other side of the lesion (Quinta, 2021).This process initiates with the re-sealing of axons in a non-regenerative stage.The structure of the axonal sub-membrane is formed by well-known cytoskeleton components.Some of these components, namely the microtubules, are involved in the formation and maintenance of axonal growth cones.
However, after a sharp depolymerization process triggered by SCI, the microtubules lose their dynamics, and the depolymerization process promotes the formation of retraction bulbs (Erturk et al., 2007; Bradke et al., 2012).This pathological process, together with the depolymerization of microfilaments,is the major contributor to the failure of axonal regeneration (Jin et al., 2009).Therefore, stabilization of microtubules and re-activation of microfilaments dynamics (polymerization and re-polymerization) could promote re-growth of the axonal shaft as well as growth cone formation and protrusion as a key step in axonal regeneration (Erturk et al., 2007; Quinta et al., 2016).
The collapse of axons that form the corticospinal tract (CST) is a result of mechanical injury where the collapse is enhanced by the secondary response that is triggered by the secretion of several proteins.These proteins have inhibitory effects on axonal growth (Kaneko et al., 2006; Quinta et al.,2014; Dominguez-Romero and Slater, 2021).One of the main proteins that participate in the secondary response is semaphorin 3A (Sema3A), which is upregulated after SCI and binds its complex receptors Neurophulin-1 and PlexinA4 (Kaneko et al., 2006; Dominguez-Romero and Slater, 2021).This process occurs at the surface of the lesioned axonal shaft and triggers an intra-axonal pathway that enhances ROS production.
The exacerbated production of ROS above physiological levels in the injured neurons within the CNS contributes to axonal regeneration failure (Hung et al.,2010, 2011).The semaphorin pathway activates the microtubule-associated monooxygenase family of proteins (MICAL), a redox-sensitive protein, and thus promotes intra-axonal ROS formation.This interaction was described by Hung and Terman (Hung and Terman, 2011) and is directly related to mitochondria-actin cytoskeleton crosstalk during cellular protrusion.This is due to intracellular levels of NADH, a mitochondrial metabolite that controls cell migration (Yin et al., 2012; van Horssen et al., 2013) via modulation of MICAL activity.Activation of MICAL promotes actin cytoskeletal destabilization and depolymerization by oxidation of filamentous actin at methionine residues, altering the formation of cell protrusions (Vanoni et al., 2013;Giridharan and Caplan, 2014).Thus, the mitochondria-controlled intracellular NAD/NADH metabolite ratio is related to the control of actin cytoskeleton dynamics, cell protrusion, and cell migration (Yadav et al., 2022).
Therapeutic Experimental Approaches
Despite several efforts in the field of neuronal repair after SCI, no effective treatments to promote functional recovery are available to date.Therefore,in this section, new insight into novel proposed therapies will be discussed(Table 1).
To achieve a successful neuronal repair after SCI, there is a series of events that must occur: (1) reseal damaged axons at the tip of injured terminals,(2) reactivate cytoskeleton dynamics, (3) transport cargo (mitochondria), (4)assemble axolemma components, and (5) functionally reconstruct axonal growth cones (Bradke et al., 2012; Lu et al., 2014; He and Jin, 2016).The critical requirement to complete this process successfully is to provide the necessary energy, i.e., ATP, which in neurons is supplied by mitochondria (Han et al., 2020).
Neurons have an exclusive property that results from their polarized structure(axons, dendrites, and cell body) with a specific function in each of these segments.The polarized structure formed among soma, dendrites, and the extension of the axons involves mitochondrial transport, as the power supply of the cell, which needs to be distributed along the entire axon to cover and maintain energy homeostasis (Sheng and Cai, 2012).Therefore, one of the proposed mechanisms to recover axonal functionality after SCI is to promote the enhancement of mitochondrial transport in damaged axons, taking into account the energy requirements critical for success (Cartoni et al., 2016; Han et al., 2016; Zhou et al., 2016).
The therapeutic approach proposed by Han et al.(2020), suggests restoring cellular energy after SCI to obtain neuronal repair.The authors establish the relationship between axonal repair and the energy needed for regeneration,proposing to enhance energy metabolism to improve axonal regrowth and reconnection after injury.They revealed that when enhancing the transport of mitochondria by genetic deletion of syntaphilin (Snph), a static anchor protein, the regeneration of cortico-spinal axons and their reconnection occurred, leading to recovered functional synapses and locomotor function(Han et al., 2020).
Moreover, the authors have established that administering creatine acted as a bioenergetics compound, facilitating regeneration and sprouting after injury.Further, they demonstrated that this outcome increased when creatine was administered in Snph-deleted mice.The authors showed that neuronal protection could be related to the maintenance of mitochondrial bioenergetics induced by creatine treatment.This was previously illustrated by Sullivan et al.(2000), who demonstrated that chronic administration of creatine ameliorated cortical damage.They showed that creatine-induced maintenance of mitochondrial bioenergetics sustained ATP levels and significantly increased ∆ψM, while ROS and Ca2+levels were significantly decreased (Sullivan et al., 2000).
Another approach to obtaining a positive therapeutic effect after SCI could be related to the use of antioxidant molecules that not only can modulate the production of oxidant molecules but also inhibit the proinflammatory macrophage activation that promotes ROS formation after SCI.Several of these molecules have been proposed as potential therapeutic approaches in the field of SCI due to their effect on neuroprotection, neuroregeneration,neuroreconnection, and neurorecovery.For example, galectin-1, a glycanbinding protein, was described as a molecule with the capacity to reduce H2O2levels in the axons after SCI.This effect prevents microfilament depolymerization and promotes F-actin repolymerization not only at the tip of the axonal growth cone but also in the filopodium of the neuronal surface,reobtaining a healthy neuronal shape and axonal regrowth (Quinta et al.,2016).
Okano’s lab described one of the main molecular mechanisms by which microfilament depolymerization occurs at the axonal growth cone after SCIinduced CST dieback (Kaneko et al., 2006).After SCI, there is a sharp increase in the expression of Sema3A, which is secreted by the meningeal fibroblast at the epicenter of the lesion (Kaneko et al., 2006).Sema3A interacts with the Nrp1-PlexinA4 receptor complex, activating a cascade of molecules that promote the collapse of all axonal growth cones and axonal degeneration.To overcome this process, Kaneko et al proposed the use of SM-216289, a small molecule from a fungal extract that can strongly inhibit the Sema3A pathway.Therefore, we proposed the inhibition of the Sema3A pathway, but our target was different.While galectin-1 interacts with the Nrp1-PlexinA4 complex receptor preventing the Sema3A binding (Quinta et al., 2014), SM-216289 binds directly on Sema3A to inhibit the binding to Nrp1 (Kaneko et al., 2006).In summary, there are different approaches to blocking the same molecular pathway in the promotion of axonal regeneration.
The experimental therapeutical approaches described so far act directly either on the mitochondrial level or at ROS formation; however, other approaches act on other cells and molecules that indirectly reduce ROS formation and therefore reduce tissue damage, demyelination, and axonal dieback.Netrin-1 protein treatment is one of these new approaches.Although the function of this protein as a diffusible guidance molecule for CST axons is well known (Meneret et al., 2017), several authors recently described its new neurorestorative function in CNS pathologies such as Parkinson’s disease (PD)and SCI.
For instance, Jasmin and colleagues demonstrated that Netrin-1 treatment,either by overexpression or by administration of recombinant Netrin-1 in the brain, causes a neuroprotective effect in the axonal structures in a murine model of PD.In this study, a PD model was created by adding a toxin that causes oxidative stress and mitochondrial dysfunction, leading to neuronal death within four weeks.Netrin-1 promoted the restoration of dopaminergic axonal projections in the striatum, suggesting a functional restorative role in the unilateral 6-OHDA (toxin) injection mouse model of PD (Jasmin et al.,2021).
A similar effect was shown by our group using recombinant Netrin-1 as a treatment for acute SCI (Quinta, 2021).Injection of recombinant Netrin-1 at the epicenter of the lesion after complete spinal cord transection produced regeneration and reconnection of descendent and ascendant axonal tracts and significant locomotor recovery.Although the mechanism by which Netrin-1 promotes these effects is not fully understood, one hypothesis could be related to the interaction of Netrin-1 and its receptors DCC and UNC5 modulating or acting like mediators in the cytoskeleton re-assembly at the axonal growth cone.This process also could involve the influx of calcium to reseal the axolemma or as a molecular mediator of the interactions between Netrin-1 and its receptors (Bradke et al., 2012; Quinta, 2022).Regarding the function of Netrin-1 on macrophage de-activation after SCI, Netrin-1 prevents the secretion of CCL2 by astrocytes after damage and therefore CCL2 cannot activate the proinflammatory macrophages.This event prevents the migration of macrophages to the epicenter of the lesion, reducing lesion volume and CST axonal dieback while preserving its myelination (Quinta, 2021).
Future Direction
Restoration of mitochondrial function after SCI seems to be an excellent target to drive new therapeutic approaches.The sealing, regeneration,and reconnection of axolemma and axoplasm after SCI depend on proper cytoskeleton repolymerization; however, this process requires biofuel production.Without ATP as a biofuel, there is no way to activate a dynamic repolymerization of microtubules and microfilaments.Moreover, high levels of ROS negatively correlate with proper actin and tubulin polymerization.Therefore, proper crosstalk between ATP formation and a decrease in ROS levels is necessary to promote neuronal repair.
A new era of manipulation of cutting-edge genetic technologies allows for the modification of certain genesin vivo.For instance, Snph deletion enhances mitochondria transport after SCI to help to recover the integrity of mitochondria and re-establish energy levels (Han et al., 2020).This effect promotes the sprouting and regeneration of CST axons as well as the recovery of locomotor function.Although this approach was successfully proved in mice, so far it has not been tested in humans; therefore, it could be an interesting approach to test in the future.Minimally invasive methods to promote axonal repair and recovery of motor function could be advantageous with respect to more complex treatment.One of these minimally invasive methods could be creatine treatment, a blood-brain barrier permeable molecule that was recently approved by FDA as a food supplement (Food and Drug Administration, 2020).
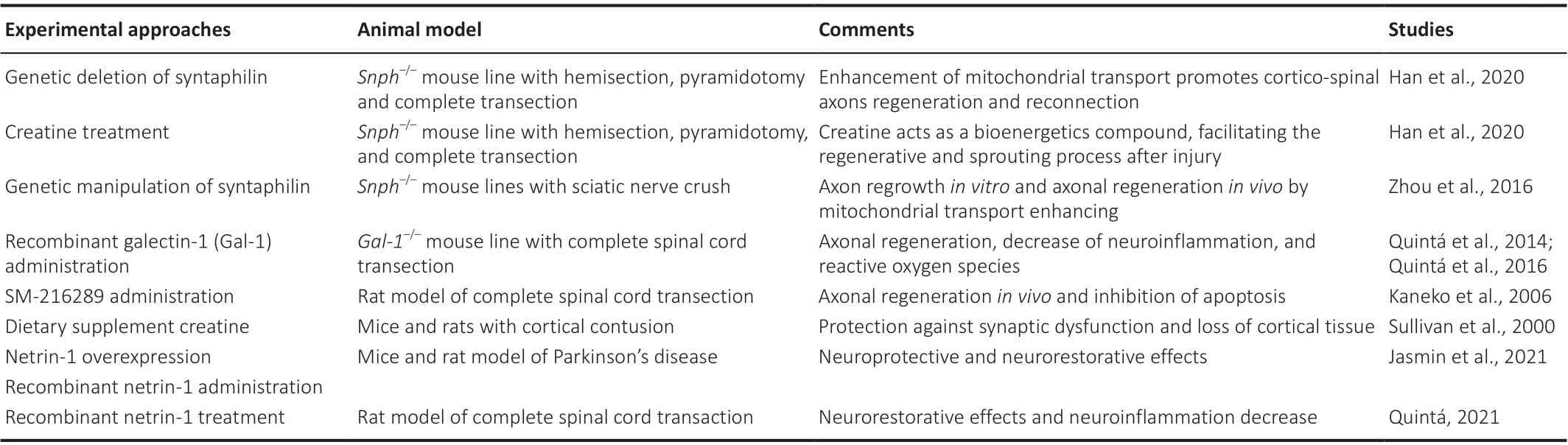
Table 1 |Novel proposed experimental approaches
As was described in this review, the administration of creatine acts as an energy facilitator to elevate the local metabolism in damaged axons in order to stimulate axonal regeneration, reconnection, and recovery of locomotion(Sullivan et al., 2000; Han et al., 2020).Therefore, this treatment could be easily implemented as a potential adjuvant in the SCI clinical setting.However,as seen in a murine model of SCI, the systemic administration of creatine had less potency regarding axonal regeneration in comparison to that observed in SnPh gene therapy, the development of new or more effective energy facilitators that more efficiently cross the blood-brain barrier could be a new direction of therapies.
Acknowledgments:I would like to thank Dr.Mariana Lagadari; Biol.Florencia Almeira and members of rehabilitation medicine department, University of Minnesota for critically reading the manuscript.
Author contributions:HRQ developed the concept of the manuscript.JS and HRQ composed and edited the manuscript.HRQ wrote the manuscript.Both authors approved the final version of the manuscript.
Conflicts of interest:The authors declare no conflicts of interest.
Data availability statement:No additional data are available.
Open access statement:This is an open access journal, and articles are distributed under the terms of the Creative Commons AttributionNonCommercial-ShareAlike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.

- 中国神经再生研究(英文版)的其它文章
- From static to dynamic: live observation of the support system after ischemic stroke by two photon-excited fluorescence laser-scanning microscopy
- MicroRNAs in mouse and rat models of experimental epilepsy and potential therapeutic targets
- The generation and properties of human cortical organoids as a disease model for malformations of cortical development
- Nanotechnology-based gene therapy as a credible tool in the treatment of Alzheimer’s disease
- Detection of Alzheimer’s disease onset using MRI and PET neuroimaging: longitudinal data analysis and machine learning
- A pancreatic player in dementia: pathological role for islet amyloid polypeptide accumulation in the brain