
槲皮素的结构修饰及生物活性研究进展
2023-03-10 01:48李阳杰曹瑞梅毛雅君邵香敏冯亚莉翟广玉
中草药 2023年5期
李阳杰,曹瑞梅,毛雅君,邵香敏,冯亚莉,翟广玉
郑州工业应用技术学院药学与化学工程学院,河南 郑州 450041
槲皮素为广泛存在于蔬菜(番茄、甘蓝、芦笋等)、水果(苹果、杏、草莓等)、中药(槐米、绞股蓝、银杏叶等)中的黄酮类化合物[1-2],具有抗氧化、抗炎、降糖、抗癌、预防和治疗心脑血管疾病等药理作用[3-10]。随着科学技术的发展,人们逐渐认识到,从天然产物中获得的化学成分应用于防病治病效果更好。因此,利用植物中的化学物质促进健康,已成为研究者广泛关注的热点[11]。
槲皮素是具有多种生物活性的天然抗氧化剂,可调节众多与疾病进展有关的细胞内、外信号通路。越来越多的研究也表明通过合成新的槲皮素衍生物可以改善槲皮素较低的溶解度和生物利用度。为了使槲皮素发挥其预防和治疗疾病的作用,尽快应用
于临床,研究学者对槲皮素进行结构修饰,主要包括对羟基的修饰生成醚和酯,对羰基的修饰生成羰基氧被取代的产物,对槲皮素A、B 环的修饰等。通过优化修饰获得了溶解性能好、生物利用度高、活性明显改善、抗癌活性增强的槲皮素衍生物[12-14],如图1所示。本文通过对槲皮素衍生物的结构修饰以及其抗癌、抗糖尿病、抗菌、抗炎、抗病毒等生物活性进行综述,并对其构效关系进行分析,为天然产物的研究、开发及利用提供参考。

图1 槲皮素的结构修饰及生物活性Fig.1 Structural modification of quercetin
1 槲皮素的结构修饰与生物活性
1.1 槲皮素抗癌衍生物的合成
1.1.1 羟基的结构修饰 前列腺癌是美国男性最常见的浸润性癌症,也是癌症相关死亡的第2 大原因。虽然有几种疗法在治疗早期取得了成功,但由于多杉醇一线治疗的耐药性问题,目前对晚期转移性前列腺癌的治疗仍然无效。Al-Jabban 等[15]采用水溶性四唑盐(water-soluble tetrazole salt,WST-1)细胞增殖实验检测了18 个合成槲皮素衍生物对人前列腺癌LNCaP、DU145、PC-3 细胞的抗增殖作用,结果表明槲皮素中C-3、C-4ʹ和C-7 羟基与短链烷基(如甲基或乙基)的烷基化略微增加了对3 种人前列腺癌细胞的抗增殖活性。然而,当支链烷基(1p 异丙基和1q 异戊基)或长链烷基(1j 丙基、1l丁基、1n 戊基、1o 己基)引入到这3 个位置时,其生物活性减弱。此外,4 种3,7-二烷基槲皮素(1b、1e、1i、1k)对3 种前列腺癌细胞的抑制率都明显高于槲皮素,其半数抑制浓度(half inhibitory concentration,IC50)是槲皮素的2~11 倍。构效关系表明:①槲皮素的C-3、C-4ʹ和C-7 羟基同时引入3 个或较长的烷基导致前列腺癌细胞的抗增殖活性显著丧失;②在槲皮素的C-3 和C-7 羟基中,化合物1b 结合2 个甲基,1e 结合2 个乙基,1i 结合2个丙基,1k 结合丁基的衍生物前列腺癌细胞的抗增殖活性更强。即3,7-O-二烷基槲皮素比3,4ʹ,7-O-三烷基槲皮素对前列腺癌细胞的抑制作用更强。
槲皮素溶于甲醇,加入无水K2CO3的N,N-二甲基甲酰胺(N,N-dimethylformamide,DMF)溶液,加入适当的卤代烷;反应混合物在室温下搅拌12~48 h,然后用乙醚-醋酸乙酯(1∶1)稀释。用盐水冲洗,有机层在无水硫酸镁上干燥,滤过后在真空中浓缩,得到相应的粗品。对粗品进行柱色谱纯化,用醋酸乙酯-正己烷(3∶7)作洗脱剂,得到化合物1a~1r,见图2。

图2 槲皮素烷基醚的合成Fig.2 Synthesis of quercetin alkyl ether
为了获得具有良好细胞毒性的新型槲皮素衍生物,Bao 等[16]使用MTT 法测定评估2a~2f 和母体化合物槲皮素对人乳腺癌MCF-7 细胞和人结肠癌Caco-2 细胞的细胞毒性。结果显示2a 和2f 对MCF-7 和Caco-2 细胞的IC50值低于槲皮素。进一步评估了2f 对人肺癌NCI-H446、A549 细胞、人胃癌MGC-803 和SGC-7901 细胞的细胞毒性,并与槲皮素进行了比较。结果表明2f 对这4 种癌细胞系均表现出强烈的细胞毒性,并且强于槲皮素,有望成为癌症化疗药物。
槲皮素溶于吡啶,加入乙酸酐反应,在丙酮中重结晶得到五乙酰槲皮素。将五乙酰槲皮素溶于18-冠-6 和CH3CN 与卤代烃(正丁基溴、烯丙基氯、肉桂酰氯、香叶基)反应,滤过,将滤液用石油醚萃取,浓缩,过色谱柱,以石油醚-醋酸乙酯(4∶6)洗脱,旋蒸,得到2a~2g,见图3。

图3 槲皮素-3,7-烷基醚的合成Fig.3 Synthesis of quercetin-3,7-alkyl ethers
为了通过化学修饰来设计具有更高效力的槲皮素衍生物,以达到治疗前列腺癌的目的。Rajaram等[17]使用WST-1 法测定24 个O-四甲基槲皮素含氮衍生物对PC-3、DU-145 和LNCaP 细胞的体外抗增殖活性。以菲斯汀、槲皮素为阳性对照,二甲基亚砜为阴性对照,结果表明3a~3x 均比槲皮素和菲斯汀具有更高的抗增殖活性。其中,在DU145 细胞中,有15 个衍生物的活性明显高于阳性对照药,衍生物3j 和3k 被确定为2 个最佳衍生物,IC50值分别为1.73、6.53 µmol/L。N,N-二戊氨基是3-O-氨基烷基-3ʹ,4ʹ,5,7-O-四甲基槲皮素抗增殖活性的最佳含氮基团。同时,24 个衍生物在抑制PC-3 和LNCaP细胞增殖方面比抑制DU145 细胞增殖更有效。其中,3j 抑制PC-3 细胞的增殖活性是槲皮素的35~182 倍,可以显著诱导PC-3 细胞凋亡,有望作为抗前列腺癌药物进一步开发。
选择3ʹ,4ʹ,5,7-O-四甲基槲皮素(3A)作为母体化合物,因为在3-OH 上连接烷基,可以在一定程度上克服槲皮素中酚羟基引起的药动学限制。3A 是以芦丁为原料,通过甲基化和糖苷水解合成。3A 通过与二溴烷烃的O-烷基化反应,得到3-O-溴代烷基-3ʹ,4ʹ,5,7-O-四甲基槲皮素(3B~3D),再与胺发生N-烷基化反应,得到24 个3-O-氨基烷基-3ʹ,4ʹ,5,7-O-四甲基槲皮素(3a~3x),见图4。

图4 槲皮素四甲基烷基氨基醚的合成Fig.4 Synthesis of quercetin tetramethyl alkyl amino ether
Mukherjee 等[18]通过在槲皮素C-3ʹ和C-5 位用不同的取代基修饰,考察10 个槲皮素衍生物脂溶性、水溶性和抗肿瘤活性。为了解决溶解性问题,在化合物4a 的C-3ʹ位选择性引入乙酸酯官能团,化合物4d 在人结直肠癌HCT116 细胞中的IC50值为3.03 µmol/L,在化合物4b 的C-5 位引入酯基,化合物4j 的IC50值为0.34 µmol/L。与槲皮素相比,在C-5 位引入酯基提高了4j 的溶解性(约为380倍)和细胞毒性(约为150 倍)。相应的酯水解物4e(IC50值为2.69 µmol/L)与其酯类4d 显示出相似的活性,细胞毒性约为槲皮素(IC50值为45.32 µmol/L)的15 倍,4k 的活性比其酯衍生物4j 降低了4 倍,而4a 的二取代酯衍生物4g(IC50值为57.5 µmol/L)和它的水解衍生物4h(IC50值为7.34 µmol/L)都失去了活性,表明C-5 和C-3ʹ的双取代可能不是很理想,C-5 或C-3ʹ与酯和相应的游离酸基团的单取代物其细胞毒性显著增加。在C-5 和C-3ʹ位引入含有弱碱基的脂肪族连接物,如N-甲基哌嗪和吡咯烷,单一取代得到4o、4p 和4v。与槲皮素相比,含吡咯烷的化合物4o 细胞毒性(IC50=1.98 µmol/L)增加22 倍,溶解度增加325 倍;含N-甲基哌嗪的化合物4p 中的C-3'位使其活性显著升高,IC50值为0.48 µmol/L,增加了90 倍,同时溶解度增加约400倍。而4p 没有显示对外周血单个核细胞的毒性,表明4p 对癌细胞有选择性毒性。4v 中C-5 位N-甲基哌嗪部分产生类似的效力,IC50值为0.55 µmol/L。
进一步研究显示,与槲皮素相比,化合物4p 在HCT116 细胞中的细胞毒性增加了96 倍。此外,在CT-26 荷瘤小鼠实验中表明,与槲皮素相比,4p 可增加荷瘤小鼠的存活率并使肿瘤体积减小(60%),说明槲皮素衍生物应用于临床具有较大的潜力。
槲皮素、溴化苄、K2CO3在DMF 溶液中反应,通过柱色谱分离出4a 和4b。4a 与1.5 当量的溴乙酸甲酯反应,得到C-3ʹ位的单乙酸酯取代物4c。4a与3.0 当量的溴乙酸甲酯反应,得到二取代物4f。化合物4c 用氢氧化钯脱苄得到化合物4d,碱作用下水解得到化合物4e。化合物4f 脱苄后得到4g,酯水解得到化合物4h。4b 与1.5 当量的溴乙酸甲酯反应,得到C-5 位的单取代物4i。4i 脱苄后得到4j,酯水解得到化合物4k。用1.5 当量的1-溴-3-氯丙烷处理4a,转化为4l,与吡咯烷和N-甲基哌嗪反应得到化合物4m 和4n。去苄基得到化合物4o 和4p。4a 用过量的1-溴-3-氯丙烷处理,转化为4q,与吡咯烷反应得到化合物4r。去苄基得到二取代产物4s。4b 用1-溴-3-氯丙烷处理,转化为4t,与N-甲基哌嗪反应得到化合物4u,去苄基得到化合物4v,见图5。

图5 槲皮素-5-或-3′-醚基乙酸衍生物的合成Fig.5 Synthesis of quercetin-5 or-3′-ether acetic acid derivatives
由于对氧化应激的不稳定性,槲皮素在细胞培养液中分解,细胞中的槲皮素浓度低于最初添加的浓度。Kim 等[19-20]将其代谢和化学敏感的羟基7-OH和3-OH 分别用新戊氧甲基(pivaloxymethyl,POM)暂时封闭,得到2 种新的槲皮素衍生物5a 和5b。其中5a 孵育后的半衰期延长至4 h,通过共聚焦活细胞成像和高效液相色谱分析表明,5a 能有效地被细胞摄取,5a 及其水解产物在细胞内的浓度可维持12 h,优于槲皮素。同时,5a 能有效被细胞摄取,其稳定性和胞内积累表现为不依赖于稳定剂的细胞抑制作用和诱导细胞凋亡。5b 比5a 更稳定,但它不能穿透细胞膜。
乙酸酐加入到槲皮素的吡啶溶液中反应得到白黄色粉末5c。硫代苯酚缓慢加入到5c 和咪唑的N-甲基吡咯烷酮(N-methyl pyrrolidone,NMP)溶液中反应得到白色粉末5d。碳酸钾和POM-碘(POM-I)添加到5d 的丙酮溶液中反应,通过硅胶柱色谱纯化(己烷-醋酸乙酯1∶1)得到黄色粉末5a。
槲皮素与二氯二苯基甲烷反应得到5e,在吡啶中与过量的Ac2O 反应得到5f。用硫酚(PhSH)、NMP 和咪唑混合物选择性除去化合物5f 7-O 位的乙酰基基团得到5g,再用苄基重新保护,得到关键中间体5h。为了选择性地在3-O 位引入POM 促进剂,用甲醇氨处理去除乙酰基,在槲皮素的C-3 和C-5 得到具有游离酚羟基的5i。羰基氧和5-OH 之间的分子内氢键阻止了5-OH 的化学反应,这导致POM-I处理时5i 在3-O 位的区域选择性烷基化。在氢解条件下,二苯甲缩酮和苄基同时脱保护,以67%的产率得到所需的5b,见图6。

图6 槲皮素-3-或-7-O-新戊氧甲基醚的合成Fig.6 Synthesis of quercetin-3-or-7-O-neopentyloxymethyl ether
Yuan 等[21]合成了15 种槲皮素衍生物,并评价其对肿瘤细胞株糖蛋白(P-glycoprotein,P-gp)、乳腺癌耐药蛋白(breast cancer resistance protein,BCRP)和多药耐药相关蛋白1(multidrug resistancerelated protein 1,MRP1)的调节活性,结果表明合成的槲皮素衍生物具有良好的P-gp 和BCRP 介导的多药耐药逆转活性。末端苯环上的甲氧基数目和O-3 侧链上的连接基类型是决定槲皮素衍生物P-gp调节活性的2 个关键结构特征。化合物6l 具有较高的P-gp 调节活性。化合物6d、6i、6l 显示出良好的BCRP 调节活性。此外,化合物6l 对P-gp 和BCRP具有等效性。由于其双重调节活性,可能是一种很好的多药耐药性(multidrug resistance,MDR)逆转剂。以上研究表明,槲皮素衍生物可作为P-gp 或BCRP 介导的癌细胞耐药安全有效的调节剂。
芦丁与甲基碘反应生成四甲基芦丁(6a),酸性水解得到6b。6b 在K2CO3存在下与2-溴乙醇回流得到化合物6c。在K2CO3存在下,槲皮素与甲基碘在DMF 中反应,得到化合物6d 和6e。6b 与3,4,5-三甲氧基苄基甲磺酸酯生成化合物6g,与2-溴-1-(3,4-二甲氧基苯基)乙酮反应得到化合物6h,与3,4,5-三甲氧基苯甲酸反应得化合物6f。中间体6c与(E)-3-(3,4,5-三甲氧基苯基)丙烯酸、3,4,5-三甲氧基苯甲酸、3,4-二甲氧基苯甲酸或4-甲氧基苯甲酸进行酯化反应,得到化合物6i~6l。为了研究含杂环的槲皮素衍生物,制备了3 个新化合物,即化合物6b 与9-(2-溴乙基)-6-氨基嘌呤、9-(2-溴乙基)-1,3-二甲基-2,6-羰基嘌呤或6-(2-溴乙氧基)-4-(3-氯-4-氟苯氨基)-7-甲氧基-喹唑啉反应,分别得到化合物6m~6o,见图7。

图7 槲皮素-3-乙基醚衍生物的合成Fig.7 Synthesis of quercetin-3-ethyl ether derivatives
为了给筛选靶向表皮生长因子受体(epidermal growth factor receptor,EGFR)的新药提供依据,Huang 等[22]合成了15 个槲皮素-3-O-氨基酸酯,采用酶联免疫吸附法测定它们对EGFR 和酪氨酸蛋白激酶(sarcoma,Src)的抑制活性,结果表明槲皮素-3-O-氨基酸酯对EGFR 激酶的抑制率<43%,而对Src激酶的抑制率高达76%。表明槲皮素-3-O-氨基酸酯对Src 激酶抑制活性比EGFR 激酶更高。因此,在槲皮素中引入氨基酸基团可以逆转EGFR 对Src 激酶的高选择性抑制作用。为了证实其生物活性,进一步测定了化合物7a~7c、7g、7k 和7m 对Src 激酶的IC50值大于50%,结果表明,6 个化合物均具有显著的抑制活性,IC50值为3.2~9.9 μmol/L。因此,为进一步开发以Src 激酶为靶点的新型抗癌药物提供了一个有前景的新型结构。
芦丁和K2CO3溶解在DMF 中,搅拌。加入苄基溴,在60 ℃搅拌,将混合物用10%醋酸调节pH为5,收集沉淀。沉淀物加到乙醇和浓盐酸溶液中,在70 ℃搅拌2 h。混合物冷却至室温,沉淀滤过并用水洗涤。粗产物用二氯甲烷-乙醇重结晶得到7。N,N-二环己基碳二亚胺(dicyclohexylcarbodiimide,DCC)、4-二甲氨基吡啶(4-dimethylaminopyridine,DMAP)、叔丁氧羰基亮氨酸、化合物7 加入四氢呋喃(tetrahydrofuran,THF)溶液中,在室温下搅拌。滤过混合物,滤液通过快速柱色谱(二氯甲烷-甲醇)纯化得到7A,加入乙醇-二恶烷混合液,10%钯碳,混合物在H2气氛中,室温下搅拌3 h。所得混合物通过硅藻土滤过,用乙醇洗涤并通过快速柱色谱纯化(二氯甲烷-甲醇),得7a。使用与7a 相同的方法,从叔丁氧羰基丙氨酸(tertbutoxycarbonylalanine-OH,Boc-Ala-OH)得到7b。相同的方法可以得到7c~7n,见图8。

图8 槲皮素-3-O-氨基酸衍生物的合成Fig.8 Synthesis of quercetin-3-O-amino acid derivatives
Kim 等[23]采用MTT 法测定槲皮素3-O 或7-O位的丙氨酸或谷氨酸衍生物(8a~8f)对HCT116 细胞、LNCaP 细胞和人包皮成纤维HS27 细胞的细胞毒性,结果表明,8a 和8b 显示出最强的逆转MDR活性(IC50=0.41、0.14 μmol/L),分别为槲皮素的20.0和58.6 倍。8c 和8d 显示出中等的调节活性(IC50=1.21、1.10 μmol/L),与槲皮素(1.92 μmol/L)相比,略有增加。8e 和8f 的MDR 逆转活性(IC50=0.78、0.71 μmol/L)分别为槲皮素的10.5、11.5 倍。8b 是逆转人子宫肉瘤MES-SA/Dx5 细胞对阿霉素耐药性最有效的药物,也能增强同一耐药细胞系中其他抗癌药的细胞毒作用,其半数有效浓度(half effective concentration,EC50)为0.8~0.9 μmol/L。8b 可抑制P-gp 的药物外排,P-gp ATP 酶分析表明8b 可与P-gp 的药物结合位点相互作用,刺激其ATP 酶活性。对8b 的理化分析表明,谷氨酸的引入可显著改善槲皮素的溶解性、稳定性和细胞摄取,使槲皮素氨基酸结合物成为安全的MDR 调节剂。
为了区域选择性地将氨基酸引入槲皮素的结构中,需要对其5 个羟基进行选择性保护。首先,槲皮素在180 ℃下与二氯二苯甲烷反应得到相应的二苯甲基酮,其余3 个羟基在吡啶中与过量的乙酸酐乙酰化,得到化合物8g。8g 在NMP 中与PhSH和咪唑反应得到7-O-单脱乙酰化产物8h。由于在碱性条件下乙酰基不稳定。因此,8h 的3,5-二乙酰基被替换为碱稳定的醚保护基团。通过顺序保护-去保护策略,用甲氧甲基(methoxymethyl,MOM)保护8h 的游离7-OH,得到关键中间体8i。将8i 的3-OH基团保护为苄基醚,然后在酸性条件下裂解7-甲氧基醚键,得到8j,再与8o 或8p 偶联,得到相应的槲皮素7-氨基甲酸酯。二苯甲基缩酮和苄基保护基同时氢解,然后使用三氟乙酸(trifluoroacetic acid,TFA)促进叔丁酯的裂解,得到8a 和8b。将8i 与(S)-N-Boc-Ala 或(S)-N-Boc-Glu(OtBu)进行1-乙基-3-[3-(二甲氨基)丙基]-碳二亚胺偶联,然后去除7-OMOM 官能团(8k),得到8l。去掉8k 和8l 的二苯基甲缩醛和叔丁酯,分别得到游离的槲皮素3-酯(8c和8d),N-Boc 保护的8k 或8j 与8o 或8p 进一步偶联,随后的去保护反应得到槲皮素7-氨基甲酰基-3-酯(8e 和8f),见图9。
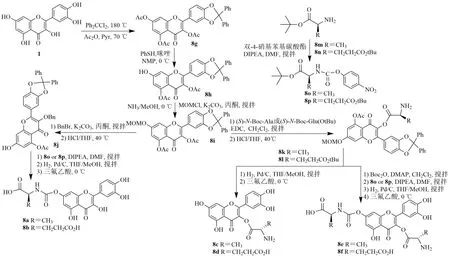
图9 槲皮素-3-O-或-7-O-氨基酸酯的合成Fig.9 Synthesis of quercetin-3-O-or-7-O-amino acid ester
Kellici 等[24-25]使用MTT 法测定槲皮素-3ʹ或-4ʹ-O-氨基酸酯类物质对DU-145、PC-3 细胞的细胞抑制活性和细胞毒活性,结果表明槲皮素-谷氨酸(9g、9o)和槲皮素-丙氨酸(9e、9m)是最有效的结合物,具有较高的细胞抑制活性。槲皮素-亮氨酸(9f、9n)和槲皮素-苯丙氨酸(9h、9p)细胞抑制活性较低。
氨基酸在密闭容器中与高氯酸和乙酸叔丁酯反应生成氨基酸叔丁酯。使用双(4-硝基苯基)碳酸酯和N,N-二异丙基乙胺(N,N-diisopropylethylamine,DIPEA)在THF 中将氨基酸叔丁酯转化为活性氨基酸甲酸酯,然后用槲皮素进行醇解。通过非水水解(TFA/二氯甲烷)实现了叔丁基保护基团的最终脱保护,得到槲皮素氨基酸酯。
将DIPEA 添加到氨基酸叔丁酯的溶液中,加入双(4-硝基苯基)碳酸酯、THF,在室温氮气环境下,搅拌12 h,加入槲皮素并搅拌反应,混合物在室温下再放置12 h。经薄层层析验证,直到槲皮素完全消失。旋蒸,粗品柱色谱,二氯甲烷-甲醇(8∶2)洗脱,洗脱液旋蒸得黄色固体化合物(9a~9d 和9i~9l)。加入二氯甲烷、TFA,在0 ℃下搅拌4 h。反应完成后,加入己烷,滤过,得黄色沉淀,用己烷洗2 次,加入二氯甲烷溶解,柱色谱,二氯甲烷-甲醇(7∶3)洗脱,旋蒸得黄色固体化合物(9e~9h和9m~9p)。所的化合物用核磁、质谱和红外光谱进行表征,可得到主要的异构体3'-O-取代的槲皮素(9a~9d、9e~9h)以及次要的异构体4'-O-取代的槲皮素(9i~9l、9m~9p),见图10。

图10 槲皮素-3′-或-4′-O-氨基酸酯的合成Fig.10 Synthesis of quercetin-3′-or-4′-O-amino acid esters
Lo 等[26]评估了槲皮素-5-O-酰基酯(10a~10g)对HCT116 细胞和人乳腺癌MDA-MB-231 细胞的抗增殖活性,及其针对1,1-二苯基-2-三硝基苯肼(1,1-diphenyl-2-picryl-hydrazyl radical,DPPH)自由基清除活性,结果表明10d、10e 和10g 具有较好的活性,其中衍生物10e 对受试癌细胞的IC50值最低,分别为(1.53±0.02)、(1.51±0.01)μmol/L,对自由基清除活性没有改变。通过比较发现长酰基衍生物比短酰基衍生物具有更好的整体活性,表明亲脂性的增加会导致生物利用度和活性的提高。一般来说,具有长酰基槲皮素衍生物比短酰基槲皮素衍生物显示出更好的抗HCT116 和MDA-MB-231 细胞的活性。5-O-酰基碳链较长的衍生物对DPPH 自由基的清除活性与槲皮素相当,而酰基碳链较短的衍生物活性略有降低。
槲皮素溶于二苯醚中,加入二苯二氯甲烷,搅拌,加入石油醚,沉淀粗产物。滤过沉淀,溶解在EtOAc 中,真空除去溶剂。所得粗产物通过快速色谱法(石油醚-EtOAc 4∶1)得到黄色固体状化合物10h。10h 和K2CO3在DMF 中搅拌溶解,室温下加入溴化苄,搅拌,所得混合物用二氯甲烷萃取。合并萃取液,真空除去溶剂。粗产物经纯化快速色谱法(石油醚-EtOAc 9∶1)得到黄色固体化合物10i。将10i 溶于三乙胺和二氯甲烷中,搅拌,加入乙酰氯,搅拌,用二氯甲烷萃取,合并有机层,真空除去溶剂,闪蒸提纯色谱得到白色固体化合物10A。相同方法可得到10B~10G。10A 和20% Pd(OH)2/C,搅拌,快速色谱法(石油醚-EtOAc 1∶1)纯化,得到黄色固体状化合物10a。相同方法可得到10b~10g,见图11。
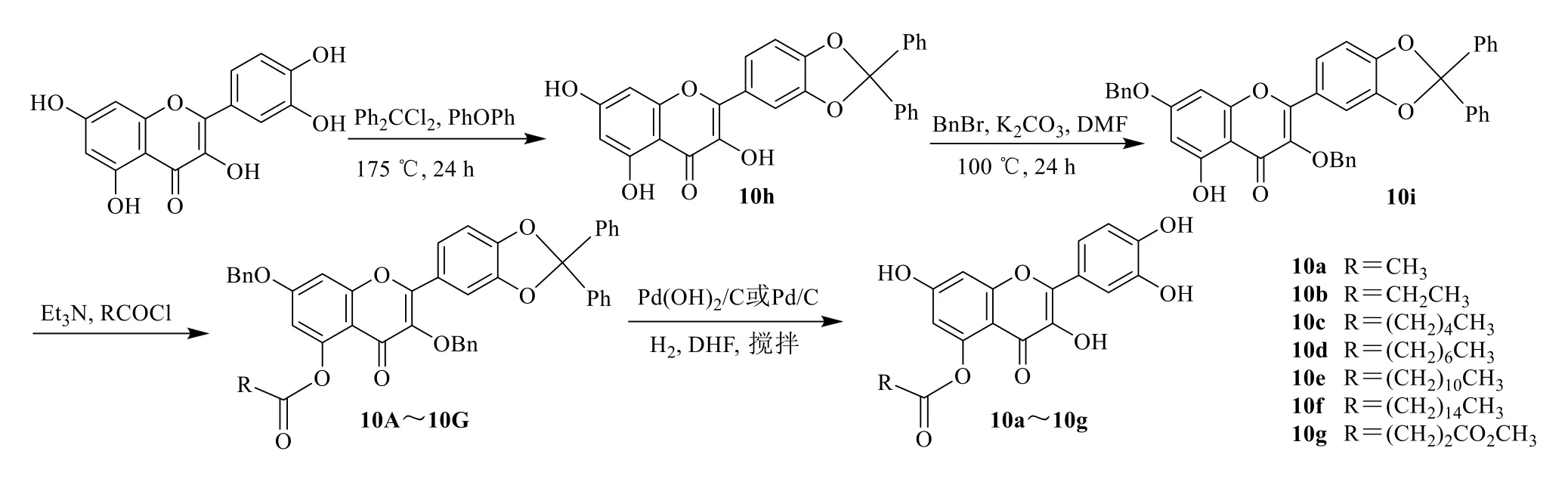
图11 槲皮素-5-酰基酯的合成Fig.11 Synthesis of quercetin-5-acyl ester
1.1.2 羰基的结构修饰 MDA-MB-231 细胞与其他类型的乳腺癌细胞不同,由于缺乏雌激素、孕激素以及EGFR2 的激素受体,故其不适合激素治疗。Adnan 等[27]使用MTT 法测定了6 个槲皮素衍生物抗肿瘤细胞增殖活性,实验显示,化合物11a 和11d都使细胞活力显著降低,分别降至43.7%和38.1%(其他化合物分别为11b 68.6%,11c 58.0%,11e、11f 无降低)。化合物11a 和11d 的IC50值分别为2.042、1.838 μmol/L,表明其具有针对三阴性乳腺癌类型的潜在抗癌活性。
槲皮素加入丙酮、K2CO3,搅拌,加入硫酸二甲酯,加热回流,滤过,热丙酮洗3 次,滤液旋蒸,甲醇重结晶,得到化合物11a。11a 溶于甲苯,加热至110 ℃,加入Lawesson 试剂 [2,4-双(4-甲氧基苯基)-1,3-二噻二膦-2,4-二硫化物],回流,将反应混合物冷却至室温,滤过,沉淀二氯甲烷溶解,柱色谱,氯仿洗脱得到化合物11b。11b 溶于乙醇中,滴加水合肼,搅拌至深绿色溶液变为橙黄色。加入胺后5 min 内通入硫化氢。0.5 h 后,停止通入硫化氢,冷水加入到反应混合物中并放于冰箱中冷藏24 h。滤过,洗涤得化合物11c。室温下11b 与过量的碘甲烷反应,反应混合物颜色从深绿色变为红色,沉淀从溶液中结晶出来,24 h 后收集,乙醚洗涤得到化合物11d。苯胺与11d 在热乙醇中反应,15 min 加入甲硫醇,产物通过硅胶柱分离,二氯甲烷-甲醇9∶1 洗脱,收集洗脱液,旋蒸得到化合物11e。将11d 悬浮在温甲醇中,加入3,4,5-三甲氧基苯胺的甲醇溶液,溶液的红色消失,反应3 h 产生黄色沉淀,沉淀物用氯仿洗涤,得到化合物11f,见图12。

图12 槲皮素羰基衍生物的合成Fig.12 Synthesis of quercetin carbonyl derivatives
Martins 等[28]应用MTT 法测定了3 个槲皮素硫或硒羰基衍生物对肿瘤细胞的生长抑制作用。对人结直肠腺癌HCT-15 细胞、MCF-7 细胞、多药耐药MCF-7-ADR 细胞、人宫颈腺癌A431 细胞活性进行测试,结果表明硒羰基衍生物12c 的细胞毒性高于其他硫族衍生物 [对于MCF-7 细胞,IC50值分别是12a>100 µmol/L,12c(3.08±1.98)µmol/L,12b、12d 无],充分证明硒羰基在细胞毒活性中的关键作用。对12c 与MCF-7 细胞的细胞毒性机制的初步研究表明,它们具有有效抑制克隆扩增的能力以及抑制硫氧蛋白还原酶活性导致细胞凋亡。与它们的羰基和硫代类似物相比,12c 显示出更高的细胞毒性,还具有克服MDR 的能力。此外,12c 对恶性肿瘤细胞具有抑制作用,而对其他细胞无毒性。与硫属元素衍生物和顺铂相比,它们表现出更高的选择性。因此,12c 可作为开发潜在的癌症化疗候选化合物。
槲皮素与硫酸二甲酯反应,得到化合物12a。12a 与五硫化二磷在THF 中反应,室温放置3 d,得到化合物12b。将12b 放入含有过量三溴化硼的二氯甲烷溶液中,室温下反应,脱去保护基,得到化合物12d。为了制备硒衍生物,使用微波,2,4-双(苯基)-1,3-二硒二膦烷-2,4-二硒化物(Woollins’试剂,WR)作为硒源[29],有效把羰基转化为硒羰基。槲皮素、乙腈、WR 在175 W 微波辐照5 min 条件下得到化合物12c。然而,尽管成功地完成了硫衍生物12b 的脱保护,得到了12d,硒衍生物12c 脱保护没有得到12e,见图13。

图13 槲皮素硫或硒羰基衍生物的合成Fig.13 Synthesis of quercetin sulfur or selenium carbonyl derivatives
1.2 槲皮素抗糖尿病衍生物的合成
金属及其配合物的药用和临床上的应用具有重要意义。Refat 等[30]通过注射链脲佐菌素诱导大鼠糖尿病模型,探讨槲皮素锌(II)(13a)配合物的抗糖尿病作用,结果表明13a 和骨髓间充质干细胞(mesenchymal stem cells,MSCs)联用可显著改善胰岛素分泌,减少细胞炎症,并有助于改善胰腺和糖代谢并发症,比单独使用MSCs 或13a 时效果更好。结果证实13a 对治疗高血糖、遗传毒性非常有效,为治疗糖尿病及其相关并发症提供了新方向。 向溶解在甲醇中的槲皮素中加入Zn(NO3)2·6H2O,加入氨水调pH 为8,室温下搅拌4 h。滤过所得黄色产物室温下缓慢蒸发过夜,然后用少量MeOH 洗涤,在干燥器中用无水CaCl2干燥得到13a,见图14。

图14 槲皮素锌配合物的合成Fig.14 Synthesis of quercetin zinc complex
Lee 等[31]报道了一系列可激活过氧化物酶受体γ 的新型取代槲皮素衍生物。部分槲皮素衍生物的过氧化物酶体增殖物激活受体激动剂活性可与目前临床使用的噻唑烷二酮类抗糖尿病药物相媲美,结果表明取代槲皮素的噻唑烷二酮衍生物,如14i~14k 表现出良好的激动活性。化合物14k 具有不受保护的槲皮素单元,是该系列化合物中活性最强的,而甲氧甲基化的槲皮素衍生物14j 与丙二酸衍生物14m 和14n 不同,其活性较低。14m 和14k 将作为抗糖尿病药物进一步研究。
槲皮素在DMF 中用过量的苄基溴,在K2CO3存在下得到14f。14f 在KH 存在下与14a 烷基化得到化合物14i。由于酚羟基需要不同的脱保护方法,因此选用甲氧甲基保护基团代替苄基保护酚羟基。以槲皮素和过量氯甲基甲醚为原料,合成了14g,并与14b 在KH 和NaI存在下偶联,合成了14j。在微量盐酸存在下,14j 在甲醇中回流,被裂解得到14k。14f 和1-溴-3-氯丙烷在KH 和NaI存在下反应得到14h。由14h 与14d 反应生成14l。14g 与14e反应生成14m。用微量的盐酸在甲醇中除去14m 的甲氧甲基保护基,得到14n。用大于5 当量的2 mol/L氢氧化锂水解14m,得到丙二酸衍生物14o。由14m在氢氧化铵存在下制备联胺14p。14l 和14m 与碘甲烷甲基化分别得到α-甲基丙二酸酯类似物14q 和14r,见图15。
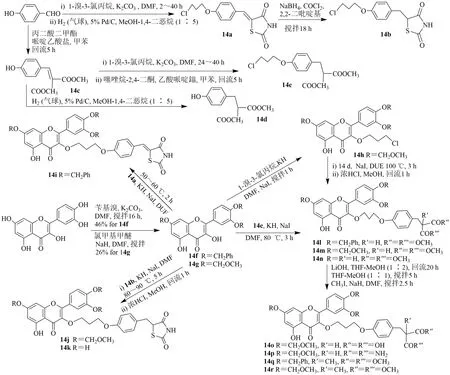
图15 槲皮素-3-丙基醚衍生物的合成Fig.15 Synthesis of quercetin-3-propyl ether derivatives
1.3 槲皮素抗菌衍生物的合成
槲皮素与金属离子配位后生物活性增加,溶解度和生物利用度增多[32]。铜是一种生物活性金属,在生物过程中具有多种作用,例如催化大量生化反应并在线粒体中运输电子起关键作用。铜(II)配合物显示出多种生物活性,可以用作抗菌、抗炎、抗肿瘤和抗病毒作用,铜(II)配合物可作为潜在的治疗药物。槲皮素具有与多种金属离子结合以增加其生物活性的能力。Moodi 等[33]使用槲皮素、乙醇胺和醋酸铜合成了槲皮素铜配合物,通过红外光谱、氢谱和碳谱图谱进行表征。光谱数据显示槲皮素3-OH 和亚胺与金属离子配位,结果表明该配合物对伯、仲醇的氧化表现出显著的催化活性,对大肠杆菌(革兰阴性菌)和金黄色葡萄球菌(革兰阳性菌)有抗菌活性。由于细胞结构的差异金黄色葡萄球菌的抗菌活性高于大肠杆菌。
槲皮素溶于乙醇,冰醋酸、乙醇胺滴加到反应混合物中。在60 ℃下搅拌回流。将所得深红色溶液浓缩冷却,得到橙色结晶沉淀用热乙醇溶液重结晶并干燥得到15a。将15a 溶解在去离子水中,加入NaOH 室温搅拌,得到橙色溶液。滴加Cu(OAc)2去离子水溶液,超声处理30 min。产生的棕色混合物后进行离心,并置于80 ℃的真空烘箱中6 h,得深橙色化合物15b,见图16。

图16 槲皮素铜配合物的合成Fig.16 Synthesis of quercetin copper complex
1.4 槲皮素抗炎衍生物的合成
Rasheed 等[34]通过研究槲皮素醋氯芬酸酯(16b)理化性质和药理作用,旨在开发无溃疡不良反应的新型非甾体抗炎药。在模拟胃液、肠液和大鼠粪便中进行水解动力学研究,测定生化指标(胃壁黏液、己糖胺)、氧化参数(全血脂过氧化物、谷胱甘肽、过氧化氢酶、超氧化物歧化酶和蛋白质含量,结果表明,合成的16b 化学稳定,生物不溶性,具有较好的亲脂性,同时保留了抗炎活性,溃疡降低,胃肠道不良反应减少。与母药相比,其蛋白质结合量小,吸收能力强,各项生化指标均表现出较好的效果。
醋氯芬酸(16x)加入氯化亚砜,室温搅拌,减压除去多余的氯化亚砜,得到黄色无定形固体醋氯芬酰氯(16a)。将芦丁加入丙酮中,加入无水碳酸钾和硫酸二甲酯,回流,滤过,减压除溶剂,产物用乙醇硫酸回流,减压脱除溶剂,乙醇重结晶得到16y。16y 溶解在含有三乙胺和4-二甲基氨基吡啶的二氯甲烷中,反应混合物冷却至-10 ℃,将16a 溶于二氯甲烷,并将其在1 h 内逐滴加入,反应混合物搅拌过夜,减压除去溶剂,乙醇重结晶获得棕色16b,见图17。

图17 槲皮素醋氯芬酸酯的合成Fig.17 Synthesis of quercetin acetyl chlorphenate
Buravlev 等[35]通过曼尼希反应合成了4 个槲皮素-8-甲基氨基衍生物(17a~17d)。结果显示,在抗坏血酸/Fe2+β 诱导的脑匀浆脂质过氧化模型中,所有衍生物均具有较高的抗氧化活性,17c 在低浓度下显示出最高的活性。小鼠红细胞氧化性溶血研究表明,具有吗啉代甲基或硫代吗啉代甲基的槲皮素衍生物具有保护原始细胞免受急性氧化应激和抗自由基活性,衍生物17c和17d在保护红细胞免受H2O2诱导的急性氧化应激的能力上超过了槲皮素。因此,17c 具有较好的抗炎作用,可用于治疗胃溃疡。1,4-二氧六环的槲皮素溶液,冰浴,加入37%甲醛水溶液,然后加入胺。将混合物升温至25 ℃,搅拌75 min(吡咯烷)或100 min(哌啶),滤过,用1,4-二氧六环洗涤干燥。在固体沉淀中加入2 mol/L HCl乙醇溶液,室温下搅拌5 min。滤过,用2 mol/L 盐酸乙醇溶液洗涤。真空中干燥,得黄色粉末产物17a或17b。将37%甲醛水溶液加入槲皮素的乙醇溶液中,加入吗啉或硫代吗啉,混合物在60 ℃加热2 h,然后冷却至室温,滤过,用乙醇洗涤干燥。在固体沉淀中加入HCl 的乙醇溶液,搅拌后混合物在水浴中加热回流,冷却至室温,滤过,用盐酸的乙醇溶液洗涤,在真空中干燥,得到黄色粉末产物(17c 或17d),见图18。

图18 槲皮素-8-甲基氨基衍生物的合成Fig.18 Synthesis of quercetin-8-methylamino derivatives
1.5 槲皮素抗病毒衍生物的合成
为了寻找具有较高抗病毒活性的化合物,Han等[36]使用芦丁和1,4-戊二烯-3-酮为原料合成了20个槲皮素衍生物,并通过半叶法评估其对烟草花叶病毒(tobacco mosaic virus,TMV)和黄瓜花叶病毒(cucumber mosaic virus,CMV)的抗病毒生物活性。结果表明20 种化合物对烟草花叶病毒显示出良好的抗病毒活性。18r 对CMV 的治疗活性高于其余化合物和宁南霉素。18k、18q~18t 表现出良好的灭活效果,优于芦丁,低于宁南霉素。由于18q~18t 对CMV 显示出良好的抑制活性,有望作为植物抗病毒剂得到进一步研究。实验发现这些衍生物的抑制活性很大程度上取决于取代基的性质,当取代基为2-呋喃基或2-噻吩基时,相应的目标化合物对CMV 和TMV 表现出良好的抑制活性。
在芦丁、K2CO3和丙酮溶液中,逐滴加入硫酸二甲酯,得到黄色胶状固体18x。将18x 加入乙醇和盐酸反应得到18y。18y 和K2CO3的混合物溶于DMF 中,加入氯醋酸乙酯反应得到黄色粉末18z;将18z 的甲醇溶液中滴加NaOH 溶液,得化合物18w;18w、DCC、DMAP 和含不同的取代基团的1,4-戊二烯-3-酮18A~18T 溶解在二氯甲烷中反应,过色谱柱,醋酸乙酯和石油醚洗脱,乙醇重结晶,得到目标化合物18a~18t,见图19。

图19 含有1,4-戊二烯-3-酮槲皮素衍生物的合成Fig.19 Synthesis of quercetin derivatives containing 1,4-pentadien-3-one
丙型肝炎病毒二酮酸(α,γ-diketoacid,DKA)的类似物或异构体通过螯合活性位点上的2 个镁离子,对丙型肝炎病毒NS5B 聚合酶有很强的抑制作用。槲皮素的抗丙型肝炎病毒(hepatitis C virus,HCV)活性部分归因于其是DKA 的结构模拟物。为了揭示槲皮素抑制HCV 活性所需的结构特征,提高抗HCV 活性,Zhong 等[37]、Lee 等[38]设计、合成了19 种槲皮素-7-O-苄基衍生物(19a~19s),并在细胞检测中对其抗HCV 性能进行评价,结果表明合成的槲皮素衍生物均表现出强效抗HCV 活性,其EC50值范围为3.8~8.7 μmol/L。在这些化合物中,间氯取代衍生物具有最高的活性(19i,EC50=3.8 μmol/L)。槲皮素在7-O 位的取代基可能类似于DKA 的间位取代基,从而增强抗HCV 活性。分子对接研究表明,槲皮素衍生物能够与2 个镁离子建立关键配位,并与丙型肝炎病毒NS5B 活性位点的残基相互作用。
槲皮素与醋酸酐反应得19x,19x 在NMP 中与硫酚和咪唑进行区域选择性脱乙酰化,得到19y。19y 与不同取代的苄基溴化物烷基化,然后用甲醇氨处理脱乙酰基,得到19a~19s,见图20。
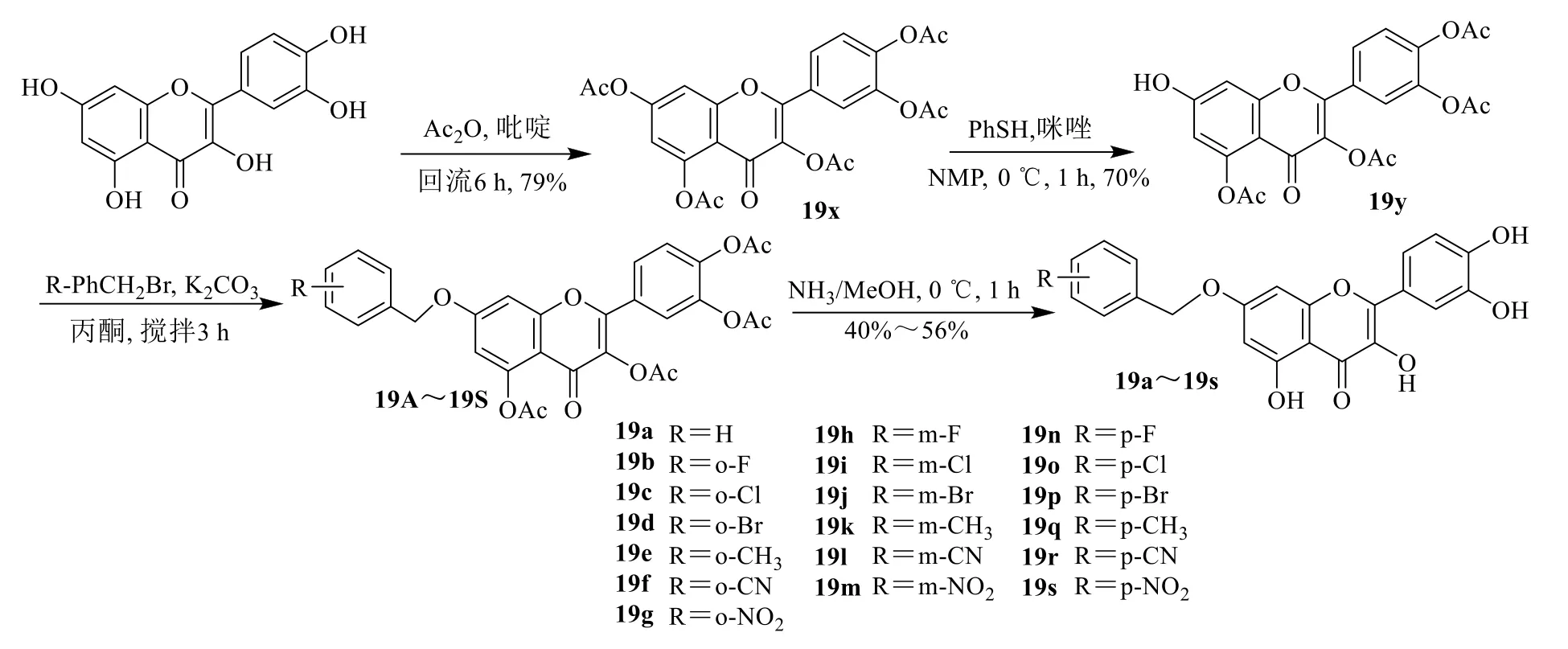
图20 槲皮素-7-O-苄基衍生物的合成Fig.20 Synthesis of quercetin-7-O-benzyl derivatives
2 构效关系
具有核心类黄酮骨架的天然产物已被证明是生物活性化合物的良好来源。大量研究表明,含有黄酮类化合物的天然产物可以克服多种MDR 细胞[39-40]。槲皮素具有C6-C3-C6碳骨架结构,由2 个苯环A 和B 组成,并通过三碳吡喃环C 连接。在槲皮素分子中,B 环存在邻二酚结构,A 环有间二酚结构,C 环有1 个烯醇式、羟基酮结构,这些结构使得槲皮素具有一些特殊的生物活性。槲皮素的3 个环是平面的,在该体系中,分子间形成3 个氢键:2 个氢键通过羰基团建立,另1 个在B 环的OH 基团之间形成。槲皮素因其显著的抗氧化和抗癌活性而被广泛研究,修饰不同基团得到的衍生物具有不同的生物活性和功效[41-42],见图21。

图21 活性较好的槲皮素衍生物Fig.21 More active quercetin derivatives
通过比较槲皮素及其衍生物的活性,可以发现
衍生物的构效关系主要取决于取代基的位置和性质。槲皮素的3ʹ-OH 是非常重要的位置,引入N-甲基-N-丙基哌嗪(4p),油水分配系数增大,水溶性提高,细胞穿透能力增强,细胞毒性活性增加了96倍,IC50值为0.48 μmol/L,在结肠癌模型中,CT-26荷瘤小鼠体内治疗17 例,与槲皮素相比,显著提高了存活率并减轻了肿瘤重量(60%)[18]。槲皮素-3ʹ-O-甘氨酸氨基甲酸酯是一种水溶性前药,仍需进一步的临床研究,以适合癌症患者使用[43]。对槲皮素3ʹ-OH 进行修饰,抗癌活性显著增强[24]。对槲皮素的3-OH 或者5-OH 进行修饰,可提高其抗病毒、抗糖尿病、抗癌和抗炎能力[20-22,26,31,36]。在槲皮素中引入羧基或酯基,可提高脂溶性、生物利用度和抗肿瘤活性[22-23];槲皮素的羰基氧被硫或硒取代,抗肿瘤活性增强[27-28];槲皮素金属配合物有较好的抗菌、抗糖尿病活性[30,33]。通过曼尼希反应,在槲皮素8位引入不同的基团,可以改善口服利用度,可开发为治疗胃溃疡的药物[35]。
3 结语
槲皮素是一种有价值的天然黄酮类化合物,由于能够调节多种靶点和信号通路而得到了广泛研究。由于槲皮素的低溶解性和生物利用度限制了其应用[44-45]。因此,设计和合成新的槲皮素衍生物来改变其局限性势在必行。目前,对槲皮素的结构进行优化修饰,已经合成了许多溶解性能好、生物利用度高的槲皮素衍生物,在抗癌、抗氧化/抗衰老、抗病毒、抗炎、降糖等方面有许多优势,应用前景广阔。希望更多的研究者积极参与其中,使天然药物经过优化修饰后早日进入临床,发挥天然药物的作用,为患者解除痛苦。
利益冲突所有作者均声明不存在利益冲突
猜你喜欢
分子催化(2022年1期)2022-11-02
化学工业与工程(2022年1期)2022-03-29
中成药(2017年9期)2017-12-19
中学生数理化·高二版(2016年3期)2016-12-26
中国塑料(2016年2期)2016-06-15
天然产物研究与开发(2016年11期)2016-06-15
合成化学(2015年10期)2016-01-17
中国病理生理杂志(2015年8期)2015-12-21
中国病理生理杂志(2015年8期)2015-12-21
应用化工(2014年11期)2014-08-16
