
基于化学模式识别筛选清炒香附的差异标志物
2023-03-10 01:48杨颜溶田瀚举李莹莹雷敬卫龚海燕谢彩侠
中草药 2023年5期
杨颜溶,田瀚举,李莹莹,贾 豪,雷敬卫*,龚海燕*,谢彩侠
1.河南中医药大学,河南 郑州 450046
2.河南省中药质量控制与评价工程技术中心,河南 郑州 450046
香附最早记载于《名医别录》[1],为莎草科植物莎草Cyperus rotundusL.的干燥根茎。《本草纲目》[2]记载:“生则上行胸膈,外达皮肤,熟则下走肝肾,外彻腰足。炒黑则止血,……青盐炒则补肾气,酒浸炒则行经络,醋浸炒则消积聚,姜汁炒则化痰饮”,表明清炒香附具有与辅料炮制香附不同的主治功效。中药炒制方法多样,包括清炒、麸炒、土炒等,香附清炒法最早见于《银海精微》,记载香附清炒法的本草古籍达61 部[3]。香附虽为“女科要药”,但其味辛而行气力强,久用易耗伤根本之气血,因此《妙一斋医学正印种子编》中岳甫嘉多为炒用[4]。关于炒制程度,历代古籍中多有提及“微炒”“炒焦”“炒黑”,用于治疗不同病症[3],明代《医学入门》[5]记载:“气病略炒”,清代《药品化义》[6]记载:“炒黑治淋漓及崩漏”,即不同清炒程度的香附确有不同主治功效。目前多为化学成分[7]、药理[8-11]、辅料炮制、药用部位以及产地等内容的研究[12-17],未见涉及清炒。
中药指纹图谱具有整体性和稳定性的特点,是评价中药质量一致性和稳定性的有效方法[18],能够对中药的主要化学成分进行宏观整体表征,层次聚类分析(hierarchical cluster analysis,HCA)、主成分分析(principal component analysis,PCA)、正交偏最小二乘法-判别分析(orthogonal partial least squares-discriminant analysis,OPLS-DA)等化学模式识别技术,能够将指纹图谱所含有的信息进行处理表达,同时避免主观加权的弊端[19]。
基于以上内容,为排除产地等方面的影响,选择河南产地香附进行分析研究,采用高效液相色谱法(HPLC),结合化学模式识别,筛选出差异标志物,对香附饮片、清炒香附、香附炭的HPLC 指纹图谱、指标成分等进行探讨,以期为清炒香附药效物质基础研究提供实验依据,为进一步完善香附质量标准提供参考。
1 仪器与材料
1.1 仪器
Agilent 1200 型高效液相色谱仪,美国Agilent公司;ME204E 型万分之一分析天平、AB135-S 型十万分之一分析天平,上海梅特勒-托利多仪器有限公司;HH-S6 型电子恒温水浴锅,巩义市予华仪器有限责任公司;KQ-700DB 型数控超声波清洗器,昆山超声仪器有限公司;FW-100 型高速万能粉碎机,北京科伟永兴仪器有限公司;101-3AB 型点热恒温鼓风干燥箱,北京中兴伟业仪器有限公司。
1.2 材料与试剂
对照品α-香附酮(批号MUST-22041007)、香附烯酮(批号MUST-22011201)、阿魏酸(批号MUST-19032928),质量分数均≥98.0%,成都曼斯特生物科技有限公司;对照品木犀草素,批号DST191020-032,质量分数≥98.0%,上海源叶生物科技有限公司;对照品对香豆酸(批号AF20041951)、5-羟基甲基糠醛(5-HMF,批号AF21030751),质量分数均≥98.0%,成都埃法生物科技有限公司;甲醇,分析纯,天津市富宇精细化工有限公司;磷酸,天津市利密欧化学试剂有限公司;娃哈哈纯净水。
1.3 药材
7 批香附药材于2021年采集于河南省新郑市大关村,经河南中医药大学陈随清教授鉴定为莎草科莎草属植物莎草C.rotundusL.的干燥根茎,燎后直接晒干,切成厚片,备用。7 批净香附饮片分别编号S1~S7。
2 方法与结果
2.1 样品制备[4]
2.1.1 清炒香附 净香附饮片(S1~S7)文火加热,炒至内部焦黄,取出,放凉,即得清炒香附(S8~S14)。
2.1.2 香附炭 净香附饮片(S1~S7)中火加热,炒至表面焦黑色,内部焦褐色,喷淋清水少许,灭尽火星,取出,放凉,即得香附炭(S15~S21)。
2.2 HPLC 指纹图谱研究
2.2.1 色谱条件 Venusil C18色谱柱(250 mm×4.6 mm,5 μm);甲醇-0.1%磷酸水溶液为流动相,进行梯度洗脱:0~12 min,5%~20%甲醇;12~30 min,20%~30%甲醇;30~40 min,30%~48%甲醇;40~52 min,48%~50%甲醇;52~55 min;50%~59%甲醇;55~25 min;59%~70%甲醇;25~80 min;70%~77%甲醇;检测波长:0~65 min,310 nm;65~80 min,254 nm;体积流量1.0 mL/min;柱温为30 ℃;进样量为10 μL。
2.2.2 供试品溶液的制备 样品粉碎,精密称取样品粉末适量,置具塞锥形瓶中,精密加入25 mL 75%甲醇,称定质量,超声处理(250 W、40 kHz)45 min,放冷,甲醇补足减失的质量,摇匀,滤过,取续滤液,经0.22 μm 微孔滤膜滤过,即得供试品溶液。
2.2.3 对照品溶液的制备 精密称取5-HMF、对香豆酸、阿魏酸、木犀草素、α-香附酮和香附烯酮对照品适量,置于5 mL 量瓶中,加入甲醇溶解,定容,分别得到质量浓度为549.3、370.7、326.7、119.3、220.0、193.3 μg/mL 的对照品溶液;分别量取6 种对照品溶液适量,配制成一定质量浓度的混合对照品溶液。
2.2.4 精密度试验 精密称取香附炭样品(S15),按“2.2.2”项下方法制备供试品溶液,再按“2.2.1”项下色谱条件进样测定6 次,以α-香附酮为参照峰(S,较于其他成分峰面积大,峰形较好,故作为参照峰),计算共有峰的相对峰面积和相对保留时间的RSD 分别为0.49%、0.16%,表明仪器精密度良好。
2.2.5 重复性试验 精密称取香附炭样品(S15),按“2.2.2”项下方法平行制备6 份供试品溶液,再按“2.2.1”项下色谱条件进样测定,以α-香附酮为参照峰,计算共有峰的相对峰面积和相对保留时间的RSD 分别为0.48%、0.37%,表明该方法重复性良好。
2.2.6 稳定性试验 精密称取香附炭样品(S15),按“2.2.2”项下方法制备供试品溶液,分别于室温下放置0、2、4、6、12、24 h,按“2.2.1”项下色谱条件进样测定,以α-香附酮为参照峰,计算共有峰的相对峰面积和相对保留时间的RSD 分别为0.49%、0.37%,表明供试品溶液在室温下放置24 h内稳定
2.2.7 指纹图谱的建立、共有峰指认、相似度评价精密称取香附样品,按“2.2.2”项下方法制备供试品溶液,再按“2.2.1”项下色谱条件进样测定,将所得数据导入《中药色谱指纹图谱相似度评价系统(2012 版)》,以S21 样品图谱为参照,采用中位数法,经多点校正、Mark 峰匹配后生成21 批样品的叠加指纹图谱和对照指纹图谱(R),结果见图1。通过与混合对照品指纹图谱(图2)比对,指认出峰3、10、11、17、21、23 分别为5-HMF、对香豆酸、阿魏酸、木犀草素、香附烯酮、α-香附酮。25 批香附样品与R 的相似度分别为0.934、0.987、0.935、0.934、0.936、0.966、0.937、0.987、0.967、0.987、0.987、0.967、0.937、0.987、0.966、0.987、0.967、0.987、0.966、0.935、0.966,均不小于0.934。
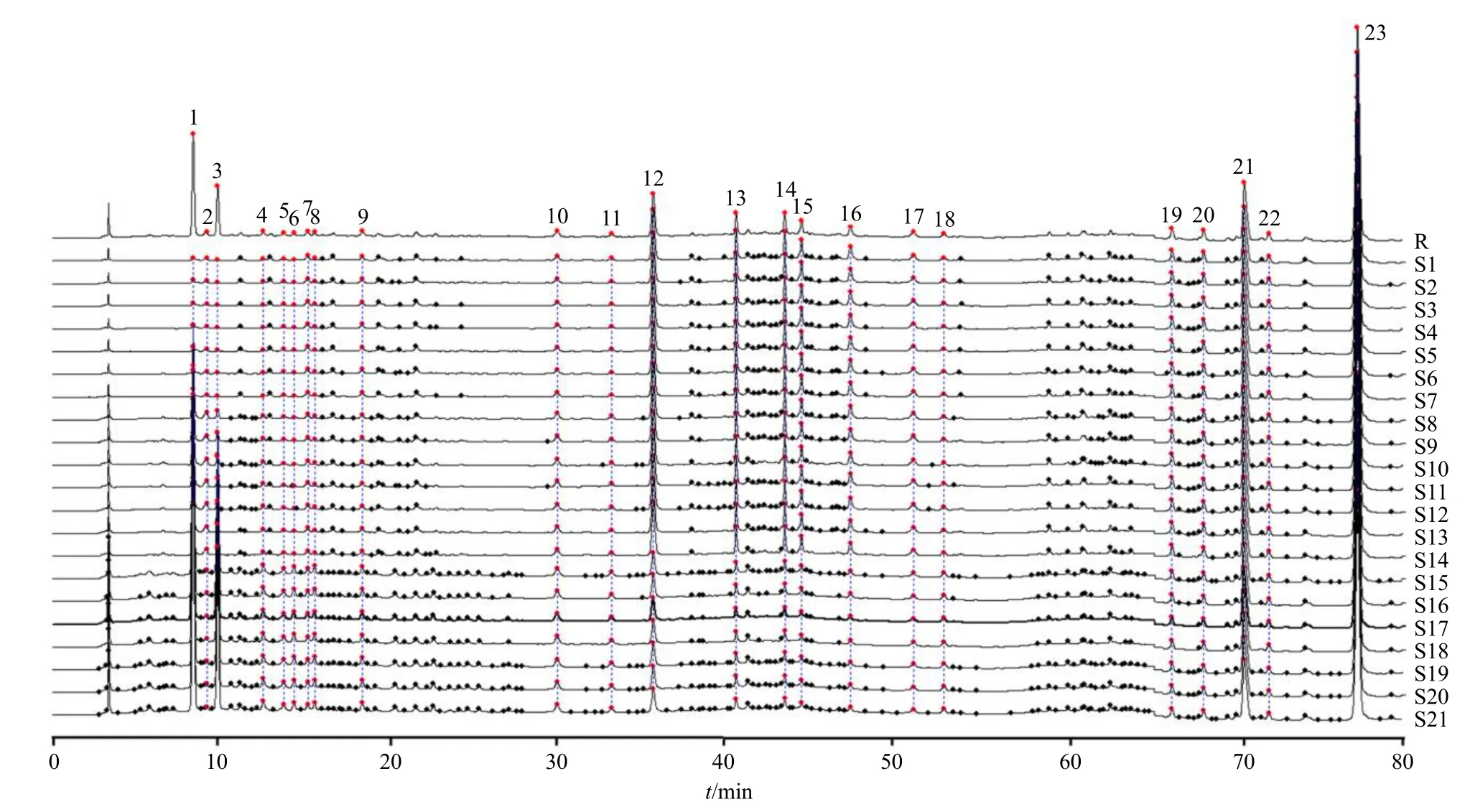
图1 21 批样品的HPLC 叠加指纹图谱及对照指纹图谱(R)Fig.1 HPLC overlay fingerprint and reference chromatogram(R)of 21 batches of samples
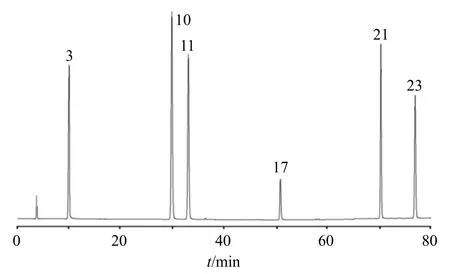
图2 混合对照品的HPLC 图Fig.2 HPLC of mixed reference substances
2.2.8 HCA 将23 个共有峰峰面积进行处理,导入SPSS.19.0 软件,采用组间连接法,以欧氏距离为分类依据,对样品进行系统聚类分析,结果见图3。可知,当组间距离为2 时,21 批样品可聚为3类:S1~S7 聚为一类,S8~S14 聚为一类;当组间距离为10 时,可聚为2 类。

图3 21 批样品的HCA 树状图Fig.3 HCA tree view of 21 batches of samples
2.2.9 PCA 采用SIMCA 14.1 软件对21 批样品中23 个共有峰进行PCA,结果见图4。结果显示21 批样品可聚为3 类:S1~S7 聚为一类,S8~S14 聚为一类。与HCA 结果一致。
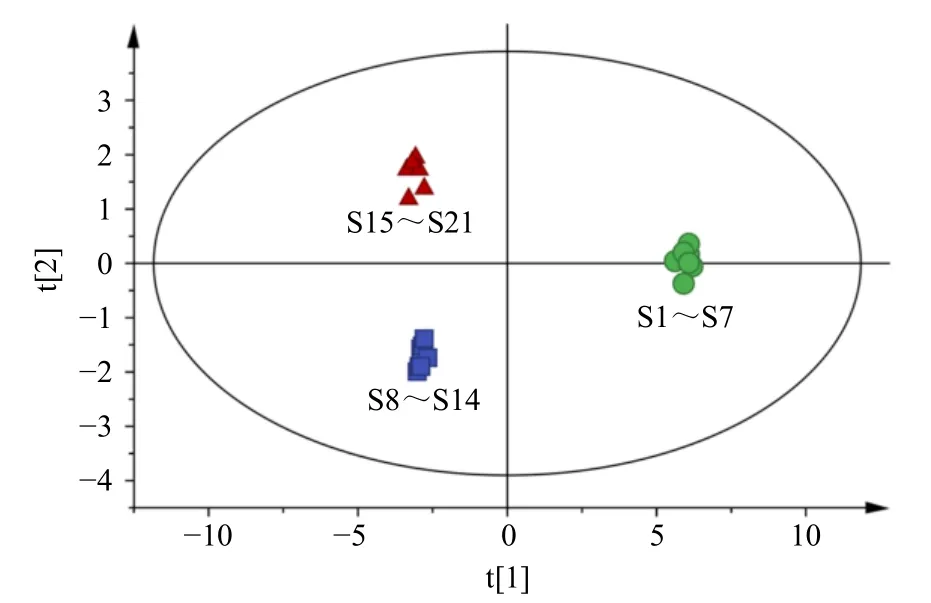
图4 21 批样品的PCA 得分图Fig.4 Score chart of PCA for 21 batches of samples
2.2.10 OPLS-DA 为进一步寻找影响不同样品间差异的成分,以23 个共有峰峰面积为变量,采用SIMCA 14.1 软件进行OPLS-DA。结果显示,R2X=0.911、R2Y=0.988、Q2=0.982 均大于0.5,表明模型拟合度较好,具有较高的稳定性与预测能力,结果见图5。可知,21 批样品可分为3 类,与HCA 结果、PCA 结果一致;为避免建立的OPLS-DA 模型出现过度拟合而影响分析结果的准确性,利用200次置换试验分别进行置换检验,结果见图6,显示置换后的R2=-0.028 8<0.3、Q2=-0.438<0.05,表明所建模型可靠,未出现过度拟合现象,可用于标志物的筛选。

图5 21 批样品的OPLS-DA 得分图Fig.5 OPLS-DA score chart for 21 batches of samples

图6 OPLS-DA 模型的200 次响应排序检验Fig.6 200-time response ranking test for OPLS-DA models
将23 个共有峰峰面积导入SIMCA 14.1 软件,对各共有峰的变量重要性投影(variable importance in projection,VIP)值进行分析,并以VIP>1 为标准筛选贡献较大的差异性成分[20],结果见图7。可知,VIP>1 的共有峰依次为2、23(α-香附酮)、17(木犀草素)、10(对香豆酸)、11(阿魏酸),提示为导致样品间差异的主要标志色谱峰。

图7 23 个共有峰的VIP 值Fig.7 VIP value of 23 shared peaks
2.3 含量测定
2.3.1 系统适用性试验 取“2.2.3”项下混合对照品溶液及“2.2.2”项下供试品溶液S10、空白溶剂(75%甲醇),按“2.2.1”项下色谱条件进样测定,记录HPLC 指纹图谱,结果见图8。可知,供试品溶液、混合对照品溶液中各色谱峰的分离度良好,空白溶剂对测定无干扰。
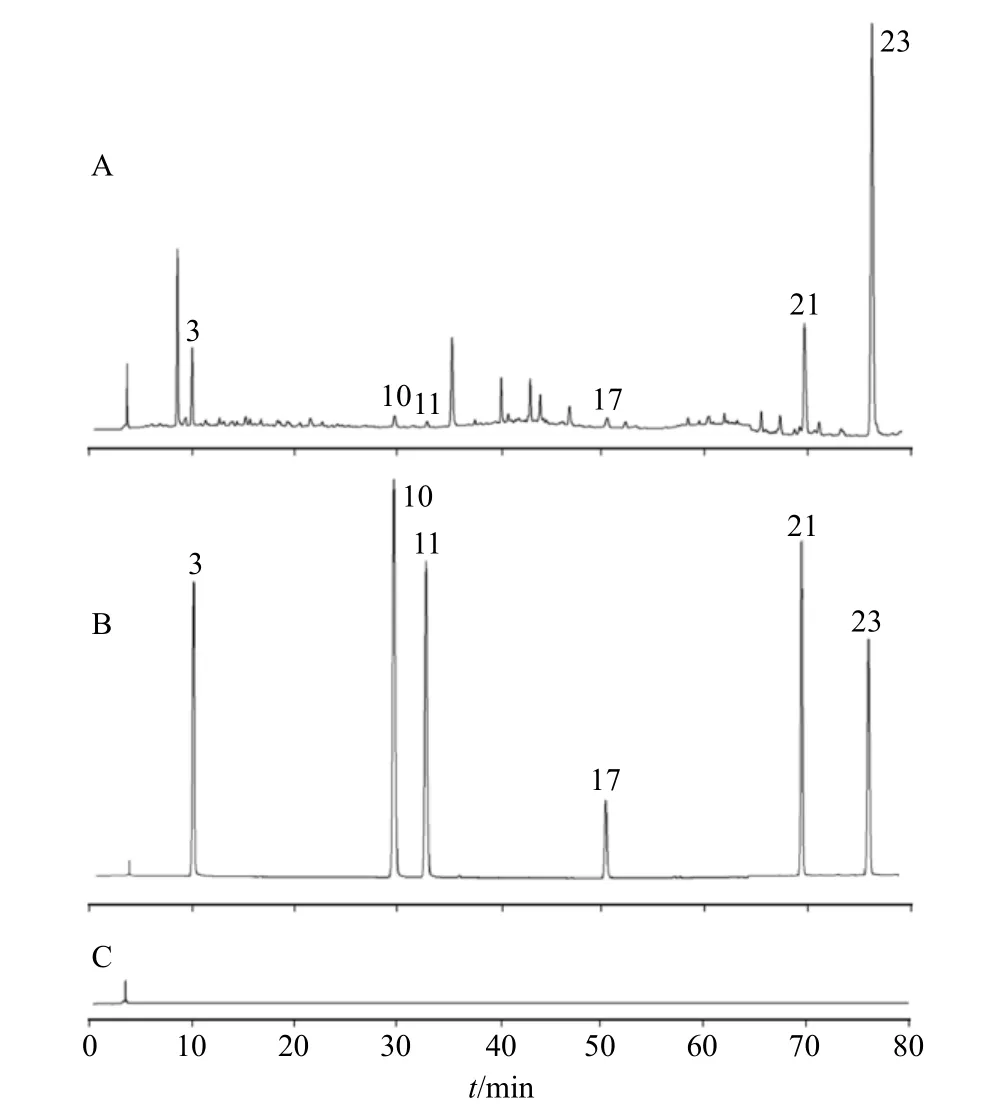
图8 供试品溶液(S10,A)、混合对照品(B)、空白溶剂(C)的HPLC 图Fig.8 HPLC fingerprint of test solution(S10,A),mixed reference substances(B)and blank solvent(C)
2.3.2 线性关系考察 精密吸取“2.2.3”项下各对照品溶液,5-HMF 对照品溶液依次稀释3.0、7.5、75.0、150.0 倍,得到质量浓度为183.1、73.2、7.3、3.7 μg/mL 的系列对照品溶液;对香豆酸对照品溶液依次稀释1.5、2.0、2.5、4.0 倍,得到质量浓度为24.7、18.5、14.8、9.3 μg/mL 的系列对照品溶液;阿魏酸对照品溶液依次稀释1.5、2.0、3.0、6.0 倍,得到质量浓度为216.7、163.4、108.9、54.5 μg/mL 的系列对照品溶液;木犀草素对照品溶液依次稀释1.5、2.0、2.5、3.0 倍,得到质量浓度为79.6、59.7、47.7、39.8 μg/mL 的系列对照品溶液;香附烯酮对照品溶液依次稀释1.2、1.3、1.5、2.0 倍,得到质量浓度为161.1、148.7、128.9、96.7 μg/mL 的系列对照品溶液;α-香附酮对照品溶液依次稀释2.5、2.8、3.0、3.5 倍,得到质量浓度为176.0、157.1、146.7、125.7 μg/mL 的系列对照品溶液。按“2.2.1”项下色谱条件进样测定,记录峰面积,以各待测成分的质量浓度为横坐标(X)、峰面积为纵坐标(Y)进行线性回归,经计算得各成分的回归方程与线性范围分别为5-HMFY=3 458.8X-0.153 1,r=1.000 0,线性范围3.7~549.3 μg/mL;对香豆酸Y=8 478.2X-1.311 0,r=0.999 6,线性范围9.3~37.1 μg/mL;阿魏酸Y=685.7X-0.107 1,r=0.999 7,线性范围54.5~326.7 μg/mL;木犀草素Y=2 404.3X+2.712 7,r=0.999 6,线性范围39.8~119.3 μg/mL;香附烯酮Y=10 102X-5.920 0,r=0.999 7,线性范围96.7~193.3 μg/mL;α-香附酮Y=31 040X+11.946,r=1.000 0,线性范围125.7~220.0 μg/mL。
2.3.3 精密度试验 取“2.2.2”项下香附炭供试品溶液(S15),按“2.2.1”项下色谱条件进样测定6次,记录5-HMF、对香豆酸、阿魏酸、木犀草素、香附烯酮、α-香附酮的峰面积,计算5-HMF、对香豆酸、阿魏酸、木犀草素、香附烯酮、α-香附酮峰面积的RSD 分别为1.48%、1.67%、0.57%、0.98%、0.51%、0.26%,表明仪器精密度良好。
2.3.4 稳定性试验 取“2.2.2”项下香附炭供试品溶液(S15),分别于室温下放置0、2、4、6、12、24 h 时按“2.2.1”项下色谱条件进样测定,记录5-HMF、对香豆酸、阿魏酸、木犀草素、香附烯酮、α-香附酮的峰面积,得5-HMF、对香豆酸、阿魏酸、木犀草素、香附烯酮、α-香附酮峰面积的RSD 分别为1.54%、0.97%、0.67%、0.16%、0.49%、0.28%,表明供试品溶液于室温下放置24 h 内稳定性良好。
2.3.5 重复性试验 取香附炭样品(S15),共6 份,按“2.2.2”项下方法制备供试品溶液,再按“2.2.1”项下色谱条件进样测定,记录5-HMF、对香豆酸、阿魏酸、木犀草素、香附烯酮、α-香附酮的峰面积,并计算含量,得5-HMF、对香豆酸、阿魏酸、木犀草素、香附烯酮、α-香附酮质量分数的RSD 分别为2.19%、1.64%、0.79%、0.26%、0.37%、0.94%,表明该方法重复性良好。
2.3.6 加样回收率试验 分别精密称取已知含量的香附炭样品(S15)9 份,分为3 组,分别按样品中成分含量的50%、100%和150%加入对应成分的单一对照品,按“2.2.2”项下方法制备供试品溶液,再按“2.2.1”项下色谱条件进样测定,记录5-HMF、对香豆酸、阿魏酸、木犀草素、香附烯酮、α-香附酮的峰面积并计算加样回收率与RSD。计算得5-HMF、对香豆酸、阿魏酸、木犀草素、香附烯酮、α-香附酮的平均加样回收率分别为 98.17%、98.64%、97.68%、98.12%、98.69%、98.57%,RSD分别为1.69%、1.89%、1.34%、1.56%、1.49%、1.83%,表明方法准确度良好。
2.3.7 样品测定 分别精密称取21 批香附样品,按“2.2.2”项下方法制备供试品溶液,再按“2.2.1”项下色谱条件进样,记录峰面积,并计算样品中各指标成分含量,对香附饮片与清炒香附、香附炭中各成分含量的平均值进行比较,结果见表1、2。由表5 可知,5-HMF 含量在香附饮片、清炒香附、香附炭中逐渐增加,炒炭后5-HMF 明显增加;对香豆酸、阿魏酸含量在香附饮片、清炒香附、香附炭中逐渐增加;清炒香附中木犀草素含量最高;香附烯酮含量在香附饮片、清炒香附、香附炭中呈下降趋势;α-香附酮在香附炭中含量最低,香附饮片经炒后α-香附酮含量略有增加。
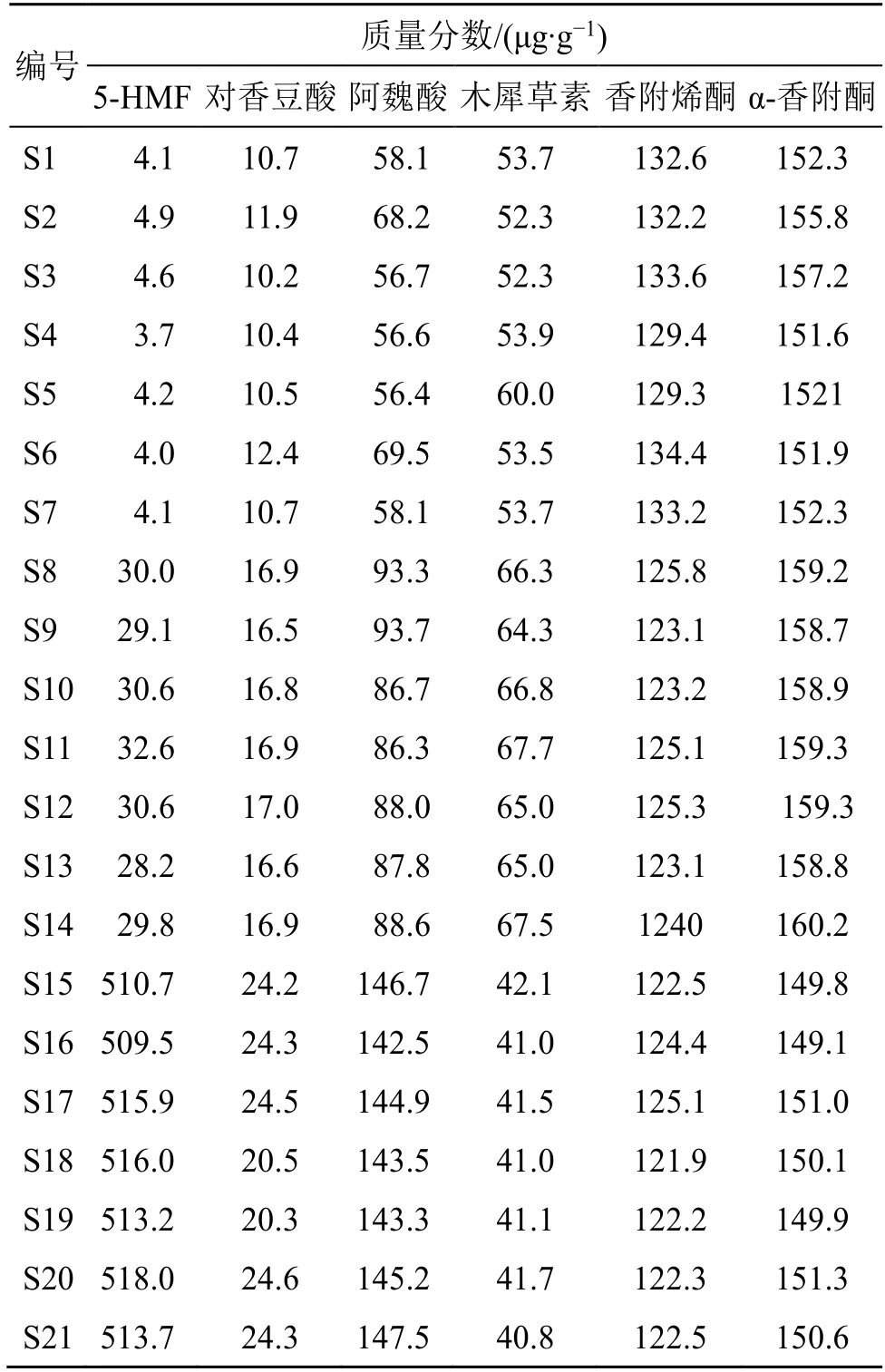
表1 21 批香附样品的质量分数测定结果Table 1 Content determination results of 21 batches of Cyperi Rhizoma
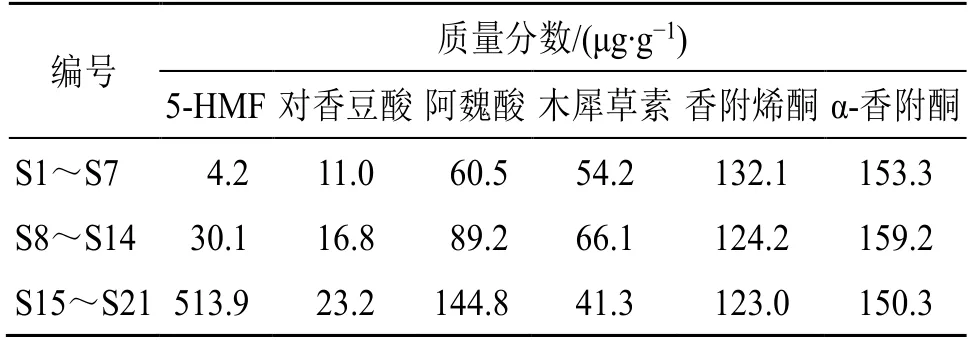
表2 各成分平均质量分数Table 2 Comparison of the average content of each ingredient
3 讨论
HPLC 指纹图谱研究结果显示,21 批样品的相似度均不小于0.934,共标定23 个共有峰,指认出3 号峰为5-HMF,10 号峰为对香豆酸,11 号峰为阿魏酸,17 号峰为木犀草素,21 号峰为香附烯酮,23号峰为α-香附酮;PCA 结果、OPLS-DA 结果与HCA结果一致,均可分为3 类,即香附饮片、清炒香附、香附炭存在一定差异,其规律符合中药炮制基本原理[21];通过VIP 法筛选出对样品影响较大的成分,筛选出未知成分2 号峰和已知成分α-香附酮、木犀草素、对香豆酸、阿魏酸为差异标志物。
含量测定结果显示,5-HMF、对香豆酸、阿魏酸、木犀草素、香附烯酮、α-香附酮的质量分数分别为3.7~518.0、10.2~24.6、56.4~147.5、40.8~67.7、121.9~134.4、149.1~160.2 μg/g;5-HMF 在香附炭中含量最高,推测与炮制程度有关[22-25];对香豆酸、阿魏酸在香附饮片中含量最少,香附炭中含量最高,研究报道含有阿魏酸的药材在体外干燥过程中会与阿魏酸松柏酯相互转化[26],推测与其有关;木犀草素在清炒香附中含量最高;α-香附酮含量经炒后略有增加,有研究报道在炮制过程中,香附烯酮转化为α-香附酮,致使其含量增加[27],推测与其有关;香附烯酮含量在香附饮片、清炒香附、香附炭中含量逐渐减少,推测与炮制过程中挥发性成分损失有关[28],提示在炮制的过程中应控制时间与温度。
综上所述,本研究筛选出未知成分2 号峰与已知成分α-香附酮、木犀草素、对香豆酸、阿魏酸为差异标志物;所建立的HPLC 指纹图谱与含量测定方法操作简单、准确,能够为清炒香附药效物质基础研究提供实验依据,可为进一步完善香附质量标准提供参考。
利益冲突所有作者均声明不存在利益冲突
猜你喜欢
食品安全导刊·中旬刊(2022年3期)2022-04-15
天然产物研究与开发(2018年2期)2018-04-04
中成药(2017年12期)2018-01-19
西江月(2017年4期)2017-11-22
小雪花·成长指南(2016年10期)2016-11-01
中国粮油学报(2016年1期)2016-02-06
化工进展(2015年3期)2015-11-11
核科学与工程(2015年2期)2015-09-26
中国烟草学报(2012年3期)2012-04-10
郑州大学学报(理学版)(2012年4期)2012-03-25
