
A new transition metal diphosphide α-MoP2 synthesized by a high-temperature and high-pressure technique
2023-02-20 13:16XiaoleiLiu刘晓磊ZhenhaiYu于振海JianfuLi李建福ZhenzhenXu徐真真ChunyinZhou周春银
Chinese Physics B 2023年1期
Xiaolei Liu(刘晓磊), Zhenhai Yu(于振海),†, Jianfu Li(李建福), Zhenzhen Xu(徐真真), Chunyin Zhou(周春银),
Zhaohui Dong(董朝辉)3, Lili Zhang(张丽丽)3, Xia Wang(王霞)4, Na Yu(余娜)4, Zhiqiang Zou(邹志强)4,Xiaoli Wang(王晓丽)2,‡, and Yanfeng Guo(郭艳峰)1,5,§
1School of Physical Science and Technology,ShanghaiTech University,Shanghai 201210,China
2School of Opto-Electronic Information Science and Technology,Yantai University,Yantai 264005,China
3Shanghai Synchrotron Radiation Facility,Shanghai Advanced Research Institute,Chinese Academy of Sciences,Shanghai 201204,China
4School of Physical Science and Technology and Analytical Instrumentation Center,School of Physical Science and Technology,ShanghaiTech University,Shanghai 201210,China
5ShanghaiTech Laboratory for Topological Physics,ShanghaiTech University,Shanghai 201210,China
Keywords: high-pressure synthesis,monoclinic MoP2,crystal structure
1. Introduction
Transition metal diphosphides (TmP2, Tm = transition metal)represent an important family with rich physical properties that have received renewed research interest owing to the nontrivial topological states associated with exotic properties such as extremely large magnetoresistance(XMR).[1–6]These fundamentally important and technically useful physical properties naturally suggest the exploration of analogue compounds;this led to the discovery of the OsGe2-type structure of TmP2.[7–9]Since such nonmagnetic TmP2generally show XMR, for example in ZrP2,α- andβ-WP2,β-MoP2,TaP2, etc., due to either the non-trivial topological electronic states or compensated carriers,[3–6]OsGe2-type MoP2is naturally expected to have similar intriguing physical properties.However, OsGe2-type MoP2has not yet been synthesized,leaving open the opportunity to achieve a complete understanding of OsGe2-type TmP2by studying the physical properties with varied Tm.
The thermodynamically stable/metastable phases of the binary Mo–P phase diagrams, including Mo3P, MoP, MoP2,and MoP4and the metastable Mo8P5and Mo4P3, have been investigated in the pressure range of 0 GPa–200 GPa[10]by first principles calculations combined with an evolutionary algorithm. Three new thermodynamically stable phases, including Mo4P(191 GPa–200 GPa),Mo2P(65 GPa–200 GPa)and Mo2P3(31 GPa–200 GPa), were predicted to exist under pressure.[10]Given that WP2has two polymorphic forms,namely,the room-temperature monoclinic(space groupC2/m) phaseα-WP2and the high-temperature orthorhombic(space groupCmc21) phaseβ-WP2,[11,12]the use of highpressure synthesis to explore new Mo–P binary phases would be valuable.
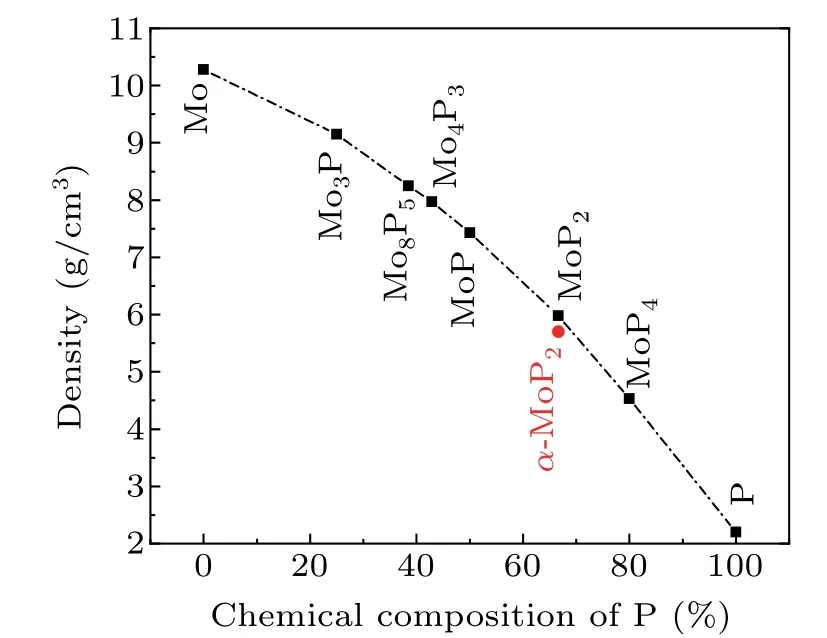
Fig.1. Densities of α-MoP2 and other Mo–P compounds. Data for the ambient pressure phases were taken from previous publications.[11,13–15]
In this paper, using high-pressure and high-temperature synthesis (HPHT) on a multi-anvil apparatus (SHTech-2000),[16,17]we succeeded in growing high-quality crystals of monoclinic MoP2, which we nameα-MoP2to distinguish it fromβ-MoP2with an orthorhombic structure. Surprisingly, the estimated structural densities of the HPHT MoP2phases are smaller than those of other phases, unlike most cases[18]seen in the plot in Fig. 1. The structural differences betweenα-MoP2and other TmP2are discussed in detail. High-pressure x-ray diffraction (HPXRD) experiments withβ-MoP2at room temperature and phonon calculations support the stability of the monoclinic phase at ambient conditions.
2. Experimental and computational methods
The staring materials, Mo (Alfa 99.7%) and red phosphorus (Katayama Chemical 200 mesh, 99%) powders, were mixed in the ratio Mo:P=1:2.5 in an Ar filled glove box.The mixture was pre-compressed into a cylinder 3.5 mm in diameter and 4 mm high,and then put into a hexagonal boron nitride chamber. The sample chamber was placed into a Ta heating furnace and then packed into a MgO octahedron. The assembly was pressed in a multi-anvil apparatus at 3 GPa–5 GPa and 1000°C–1200°C for 30 min. Detailed information about the HPHT synthesis technique can be found elsewhere.[16]
Examinations of the crystallographic phase and crystal quality of the product were performed with a Bruker D8 single-crystal x-ray diffractometer with MoKα(λ=0.71073 ˚A) at 298.0 K. Using Olex2,[19]the structure was solved with the SHELXT[20]structure solution program and was refined with the SHELXL[21]refinement package using the least squares minimization. The parameters for data collection and details of the structure refinements are given in Table 1. Atomic coordinates and thermal displacement parameters(Ueq)are given in Table 2, anisotropic displacement parameters are given in Table 3 and selected interatomic distances are given in Table 4.
To examine whetherβ-MoP2could be pressed into theα-phase at high pressure and ambient temperature, we performed a HPXRD experiment. Measurements onβ-MoP2crystals were carried out in a symmetric type diamond anvil cell with a diamond culet size of 300 mm. Silicon oil was used as the pressure transmitting medium[22]and the pressure was measured with the ruby fluorescence technique.[23]Highpressure synchrotron XRD patterns were collected at beamline BL15U1 of the Shanghai Synchrotron Radiation Facility,with a monochromatic beam wavelength of 0.6199 ˚A, using the MarCCD detector. The Fit2D software package was used to process the XRD data.[24]The XRD patterns ofβ-MoP2were analyzed with Rietveld refinement using the GSAS program package[25]with the user interface EXPGUI.[26]
The total energy calculations and structural optimization were performed using the plane-wave basis, projected augmented wave (PAW) potentials, and generalized gradient approximation with the Perdew–Burke–Ernzerhof exchange–correlation functional as implemented in the Viennaab initiosimulation package.[27–31]The frozen-core all-electron PAW potentials were used with 4p65s14d5and 3s23p3treated as valence electrons for Mo and P, respectively. The cutoff energy (900 eV) for the expansion of the wave function into plane waves and Monkhorst–Pack[32]k-meshes (k-point density 0.02 ˚A-1) were chosen to ensure that all the enthalpy calculations are well converged to better than 1 meV/atom.Phonon calculations were performed on the structures to determine their dynamic stability,by using a finite displacement approach as implemented in the PHONOPY code.[33]

Table 1. Crystal information and structure refinement results for α-MoP2 at 298.0 K.
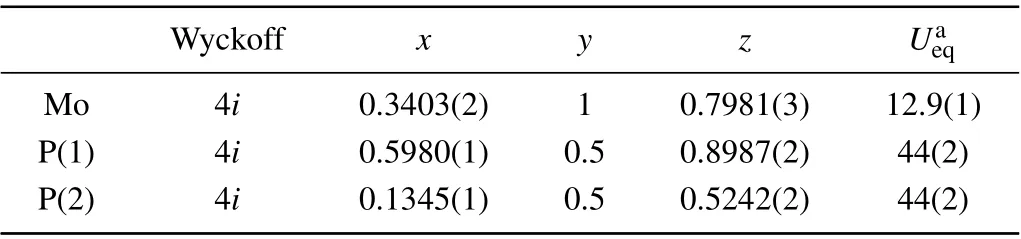
Table 2. Atomic coordinates and equivalent isotropic displacement parameters(˚A2×103)for α-MoP2.
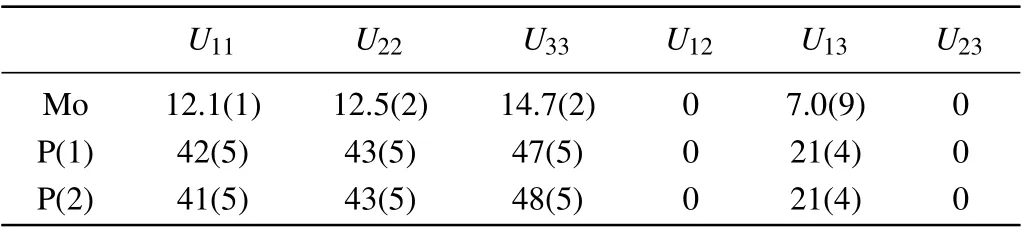
Table 3. Anisotropic displacement parameters(˚A2×103)for α-MoP2.
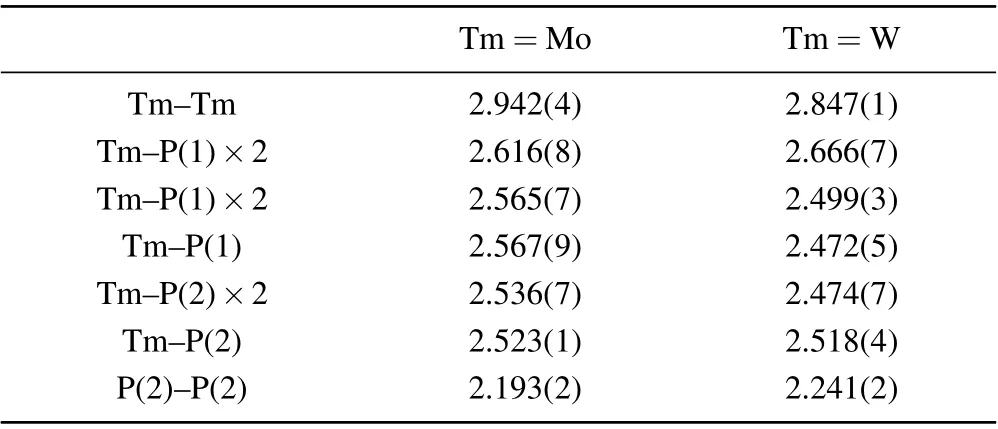
Table 4. Selected interatomic distances (˚A) for OsGe2-type TmP2 (Tm=Mo/W).
3. Results and discussion
The sample assembly for the preparation ofα-MoP2was first precompressed under a pressure of 4 GPa and then heated up to 1100°C within 20 min while maintaining the pressure. After reaction for 2 h at this temperature, the sample was slowly cooled down to 900°C within 3 h, followed by quenching to room temperature and finally releasing the pressure. The stoichiometric ratio of the sample was examined by element-resolved energy dispersive x-ray spectroscopy, as shown in Fig.2(b). Average compositions measured at different points on the crystal reveal good stoichiometry with the atomic ratio of Mo:P (33.51%:66.49%)=1:2. The analysis of single-crystal XRD data showed thatα-MoP2crystallized in theC2/mspace group with the lattice parameters ofa=8.7248 ˚A,b=3.2322 ˚A,c=7.4724 ˚A andβ=119.263°.The OsGe2-type structure of MoP2is different from the orthorhombic phase,[12]which was synthesized for the first time. The diffraction patterns in the reciprocal space along the(hk0),(h0l),and(0kl)directions are shown in Figs.2(c)–2(e),demonstrating the high quality of the crystal by the perfect reciprocal space reflections without any other miscellaneous points. Information about the crystal and the results of structural refinement are summarized in Table 1.
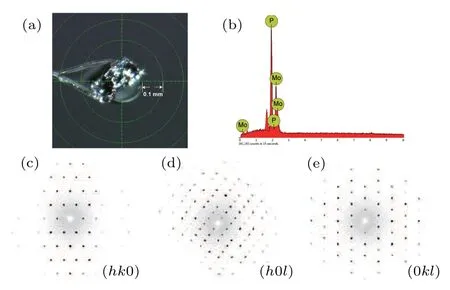
Fig.2. (a) Optical image of α-MoP2 single crystal. (b) Energy dispersive x-ray spectroscopy of α-MoP2 shows that the atomic ratio of Mo:P=1:2.(c)–(e) Single-crystal XRD patterns in reciprocal space along the (hk0),(h0l),and(0kl)directions.
It is necessary to compare the two crystallographic phases of MoP2for a better understanding of the structure ofα-MoP2.α-andβ-MoP2were grown by the HPHT and chemical vapor transport methods,respectively. Orthorhombic MoP2crystallizes in the space groupCmc21, with one Mo and two P ions located on 4aWyckoff positions and chains of molybdenum polyhedra running along the crystallographicaaxis. Monoclinic MoP2crystallizes in the space groupC2/m, with one Mo and two P ions located on 4iWyckoff positions and chains of molybdenum polyhedra running along the crystallographicbaxis. Sketches of the crystal structures of both phases are illustrated in Fig. 3. One of the roles of high pressure is to increase the coordination number (CN) of the atoms. As expected, monoclinic MoP2has a higher CN (8 for Mo) than orthorhombic MoP2(7 for Mo). Along with the higher CN,the interatomic distances are larger. The shortest Mo–P distance inα-MoP2is 2.52 ˚A, which is larger than the value of 2.48 ˚A inβ-MoP2. Moreover,the average of eight Mo–P distances inα-MoP2is 2.57 ˚A,which is also larger than the value of 2.49 ˚A inβ-MoP2.
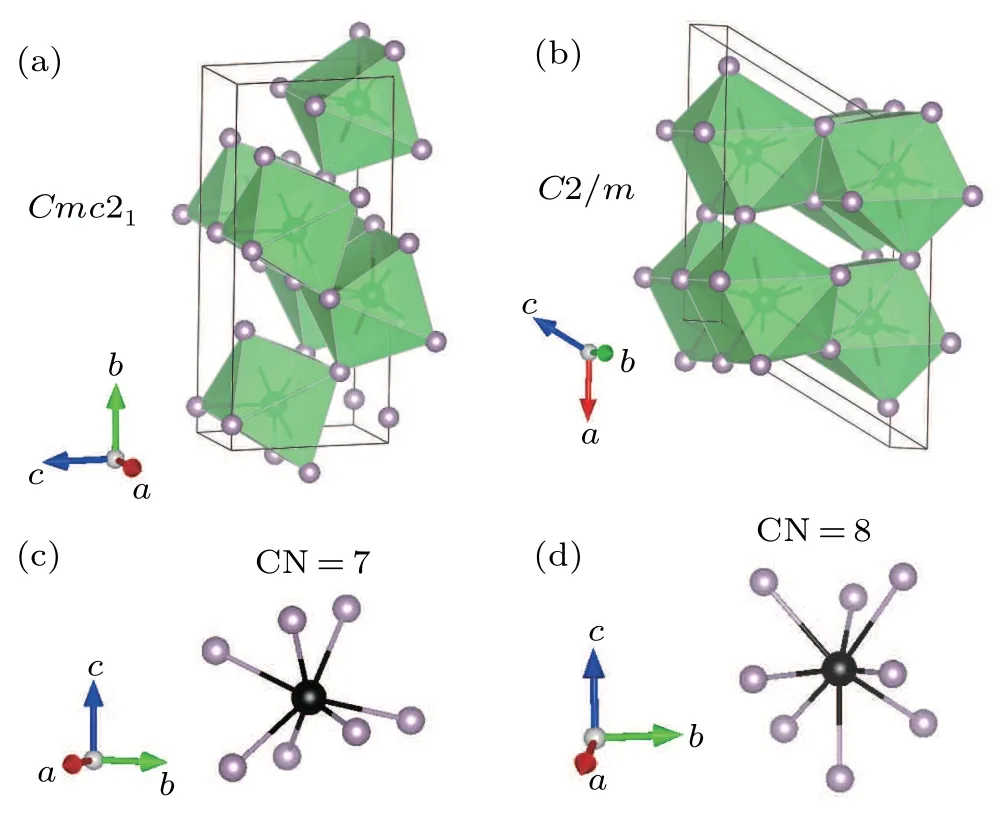
Fig.3. Schematic crystal structure and the CNs for Mo atoms of(a)β-MoP2 and(b)α-MoP2.
In most cases, single-crystal growth for the same phase can be achieved by a variety of techniques. For example, the CrP4crystals could be prepared by both HPHT and the Sn flux method under ambient pressure.[15,34]However,for some phases, they can only appear under extreme conditions such as HPHT. Synthesis ofα-MoP2has not yet been achieved under ambient pressure, maybe because the chemical bonds among the elements cannot be formed, while HPHT conditions provide an environment facilitating the formation ofα-MoP2rather than pressing a denser phase. From the viewpoint of crystallography, orthorhombicβ-MoP2has a higher structural symmetry than monoclinicα-MoP2. Materials with a higher structural symmetry tend to crystallize into closepacked structures with larger densities, which might account for the smaller structural density ofα-MoP2.
Since metal elements of the same group have similar physical properties and larger radii with increasing atomic number, intermetallic compounds of the same structure generally change steadily in volume with the substitution of metal elements in the same group. Figure 4 shows the lattice parameters forα-TmP2as a function of atomic number, including the axes, beta angle and unit cell volume. It is apparent that there is no simple positive evolution between the lattice parameters and the atomic number. According to a simple estimation, WP2should have longer axes and a larger volume than MoP2,which is contrary to the observations in Figs.4(a)and 4(b). To see the detailed near-neighbor environments in MoP2and WP2,the adjacent bond lengths and bond angles of Mo and W are depicted in Fig. 5. The bond lengths between Tm and P atoms in MoP2are all slightly smaller than those in WP2, except for the bond lengths of the two Tm–P(1). It is worth noting that if Mo is replaced by W,a similar method to a change in external pressure,[35]not only is the metal atom distance reduced but the angle ∠P(2)–Tm–P(2) is increased.This change is hypothesized to be due to the nonlinear extraction behavior shown in Fig. 4, which arises from the effect of the lanthanide contraction that could effectively alter the interatomic space. The distance between Mo metal atoms is 2.942 ˚A, which is reduced to 2.847 ˚A in WP2. Meanwhile,∠P(2)–Tm–P(2) is increased from 65.05°in MoP2to 68.92°in WP2. Therefore, lanthanide contraction can be applied to control the bond length or strength by simulating the external pressure.
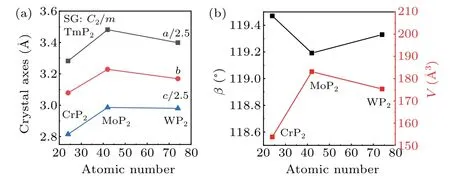
Fig.4. Lattice parameters a, b, c, β, and volume V in α-TmP2 (Tm=Cr,Mo,W).SG denotes the space group.
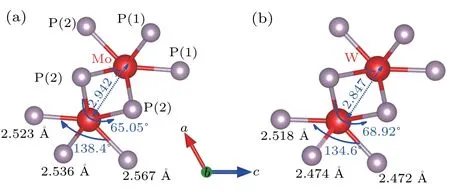
Fig.5. Nearest-neighbor environments in the α-MoP2 (a) and α-WP2 (b)structures.
It should be noted that the HPHT phaseα-MoP2is quenchable and stable at ambient conditions. Therefore,to establish the temperature–pressure phase diagram for MoP2,we also tried to synthesize monoclinic MoP2by applying a larger pressure on the orthorhombic phase at room temperature. The HP-XRD measurements onβ-MoP2up to 30 GPa gives the evolution of lattice parameters as a function of pressure seen in Fig.6(a). It is apparent that all diffraction peaks shift towards higher angles without structural phase change with increasing pressure,consistent with the results of the previous experiment measured from ambient conditions to 6.8 GPa and 839 K[36]The enthalpies of theCmc21andC2/mstructures are plotted as a function of pressure in Fig.6(b),which shows clearly that theCmc21structure is the most stable up to 40 GPa, implying that the application of pressure alone could not achieve theC2/mphase. The results are in excellent agreement with the experiment. Besides,we calculated the phonon dispersion curve for theC2/mphase at 0 GPa using the supercell method,which shows no imaginary phonon frequencies in the whole Brillouin zone, seen in Fig. 6(c), indicating that theC2/mstructure is dynamically stable at ambient conditions.
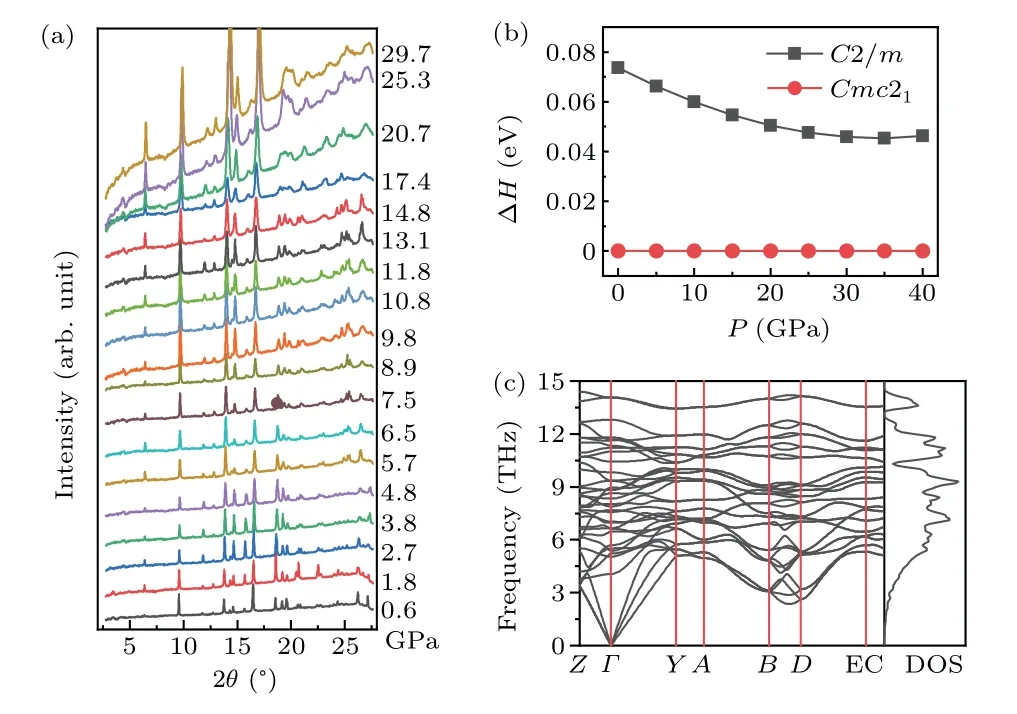
Fig.6. (a)The selected angle dispersive XRD patterns of β-MoP2 under various pressures up to 29.7 GPa at room temperature. (b)Enthalpy curves for C2/m and Cmc21 structures as a function of pressure. (c)Phonon dispersion and phonon densities of states(DOS)curves for the α-MoP2 at 0 GPa.
4. Conclusion
In summary, using a multi-anvil HPHT synthesis technique we have discovered a new high-pressure phase,α-MoP2,with aC2/mstructure.α-MoP2has a larger CN and an anomalous smaller structural density as compared with those of the ambient pressure phaseβ-MoP2, which is mainly due to lanthanide contraction. The total energy and phonon calculations indicate thatα-MoP2is dynamically stable at ambient conditions and could not be obtained by pressingβ-MoP2at room temperature. Our results provide a new phase of MoP2and would be a good guideline for the synthesis of novel transition metal phosphides.
Acknowledgments
Project supported by the National Natural Science Foundation of China (Grant Nos. 92065201, 11874264, and 11974154)and the Starting Grant of ShanghaiTech University and Analytical Instrumentation Center, SPST, ShanghaiTech University (Grant No. SPST-AIC10112914). Dr. X. L. Wang acknowledges support from the Natural Science Foundation of Shandong Province,China(Grant No.ZR2022MA004).
猜你喜欢
小学生学习指导(中年级)(2021年11期)2021-11-30
延河(下半月)(2021年7期)2021-08-10
恋爱婚姻家庭(2019年7期)2019-07-03
海峡姐妹(2019年4期)2019-06-18
恋爱婚姻家庭(2019年19期)2019-01-28
中华家教(2018年9期)2018-10-19
中华奇石(2014年12期)2014-07-09
当代(2014年2期)2014-05-20
当代(2014年2期)2014-05-20

- Chinese Physics B的其它文章
- The coupled deep neural networks for coupling of the Stokes and Darcy–Forchheimer problems
- Anomalous diffusion in branched elliptical structure
- Inhibitory effect induced by fractional Gaussian noise in neuronal system
- Enhancement of electron–positron pairs in combined potential wells with linear chirp frequency
- Enhancement of charging performance of quantum battery via quantum coherence of bath
- Improving the teleportation of quantum Fisher information under non-Markovian environment