
靶向GPCRs的药物筛选方法
2023-02-18 02:30孟庆余高永静
中国医药生物技术 2023年1期
孟庆余,高永静
G 蛋白偶联受体(G-protein coupled receptor,GPCR)是一类 7-次跨膜蛋白,参与介导多种重要的生理功能,是畅销药物中占比最高和最成功的靶点之一。几十年来,靶向GPCR 的药物研发一直是学术界和制药界的热门焦点,这些已发现并批准使用的 GPCR 药物为多种人类疾病的治疗提供了可能性,包括癌症、病毒感染、炎症性疾病和代谢性疾病等,例如艾塞那肽、利拉鲁肽、利西拉肽和阿比鲁肽等靶向胰高血糖素样肽 1 受体的肽类药物已用于 2 型糖尿病的治疗[1];靶向钙敏感受体的变构调节剂西那卡塞已批准用于甲状旁腺功能亢进治疗[2]。
目前,一些基于荧光或化学发光检测的功能性分析试剂盒已商品化,便于 GPCR 药物筛选开发。然而,就研发新药而言,现有技术的高投资和低产出的矛盾凸显出对更高效的药物筛选方法的需求。传统的 GPCR 药物筛选技术通常是基于放射性的竞争结合分析,不仅价格昂贵而且有害健康。目前,尽管已经有多种靶向 GPCR 药物开发成功的案例,但是该类药物多数仍呈现出一定的毒副作用,并且尚有超过 100 种孤儿 GPCRs(oGPCRs)的配体未知。因此,开发新的靶向 GPCR 的药物需要更经济高效的药物筛选方法,这无疑是未来药物发现工作的新挑战。在过去几十年中,为了发现更精准和全面的 GPCR 靶向药物,人们一直致力于开发基于细胞水平的信号依赖性的分析方法,这些策略的开发为药物发现提供了重要平台。近年来,由于受体药理学的进展、结构生物学的突破和生物技术的创新,GPCR药物发现的新途径相继出现,包括高内涵成像技术、3D 晶体学药理分析技术以及虚拟筛选等。在多种靶向 GPCR 药物高通量筛选(high throughput screening,HTS)的技术中选择合理的筛选策略对于获得高效、高特异性和低毒性的靶向药物至关重要。
本综述总结了使用最广泛的 GPCR 检测方法及其优缺点以及用于 GPCR 药物发现的 HTS 技术的最新进展。
1 GPCRs 及其信号通路
GPCRs 也称为 7-次跨膜螺旋(7TM)受体,是人类膜蛋白中一类最大的受体超家族,大约 800 个成员,可感知包括光、气味、味道、激素和神经递质在内的多种细胞外信号[3],在生理学和病理学方面发挥关键的功能。研究表明,大约 40% 的临床批准药物靶向 GPCRs[4]。因此,了解GPCR 的信号机制对于更好地开发 GPCR 靶向药物至关重要。
GPCR 通过与异源三聚体鸟嘌呤核苷酸结合调节蛋白(G 蛋白)偶联[5]。异源三聚体 G 蛋白由 α、β 和 γ 亚基组成,在基础状态下,Gα 被二磷酸鸟苷(GDP)占据,激动剂激活受体后,导致 GDP 与三磷酸鸟苷(GTP)交换,随后 Gα 亚单位与 Gβγ 亚单位分离,分别与下游效应蛋白相互作用,继而引发一系列级联反应,完成跨膜信号转导[6]。根据其下游功能和序列,Gα 亚基分为四个家族:Gαs(Gαs和 Gαolf)、Gαi/o(Gαi1、Gαi2、Gαi3、Gαo、Gαt1、Gαt2、Gαt3 和 Gαz)、Gαq/11(Gαq、Gα11、Gα14 和 Gα16)和Gα12/13(Gα12 和 Gα13)[7](图1)。一个受体可与多个G 蛋白亚型偶联。
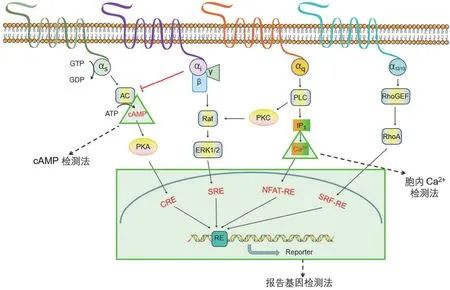
图1 GPCRs 偶联不同类型 G 蛋白信号通路示意图(Gα 亚基可分为 Gαs、Gαi/o、Gαq/11 和 Gα12/13,在配体结合后,GPCR 改变其构象并激活偶联的 G 蛋白,随后通过下游效应器促进相应第二信使和报告基因的产生。图中红色字体表示常用 HTS 分析的检测点,包括 cAMP 检测、Ca2+ 通道检测和报告基因分析)
GPCR 的典型信号通路是通过激活位于细胞表面的受体,将细胞外化学信号传递到胞内。目前的研究表明,GPCRs主要通过两条途径介导和调节生理功能:G 蛋白途径和β-arrestin 途径。传统 GPCR 激动剂与受体结合后激活 G蛋白信号通路,而 β-arrestin 偏好配体则主要激活β-arrestin 通路[8-9]。Gαs 和 Gαi/o 蛋白偶联信号通路作用于腺苷酸环化酶(AC)从而催化 ATP 转化为环腺苷酸(cyclic adenosine monophosphate,cAMP)。与 Gαs 蛋白偶联的受体直接激活该过程,促进 cAMP 的产生;而与 Gαi/o蛋白偶联的受体则抑制 AC 产生 cAMP。因此,与 Gαs 结合的 GPCR 可以抵消与 Gαi/o 结合的 GPCR 产生的效应,反之亦然。细胞质中 cAMP 的含量对生理病理条件下的多种离子通道的开放和丝氨酸/苏氨酸特异性蛋白激酶 A(PKA)家族成员的激活状态起着关键的作用。cAMP 被认为是第二信使,PKA 是第二效应器[10-11]。Gαq/11 通路的效应物是磷脂酶 C-β(PLCβ),它催化膜结合的 4,5-二磷酸磷脂酰肌醇(PIP2)裂解为 1,4,5-三磷酸肌醇(IP3)和二酰基甘油(DAG),IP3 作用于内质网(ER)膜上的 IP3 受体,促进内质网释放 Ca2+,而 DAG 沿质膜扩散并激活膜的丝氨酸/苏氨酸特异性蛋白激酶 C(PKC)结合部分[12-14]。
β-arrestin 是细胞中重要的调节蛋白,包括 β-arrestin 1和 β-arrestin 2,在持续激动剂刺激的情况下,GPCR 被特异性 GPCR 激酶(G protein-coupled receptor kinases,GRK)磷酸化,β-arrestins 招募到磷酸化 GPCR 使受体与 G 蛋白解偶联,并将受体靶向网格蛋白内吞囊泡,最终终止 G蛋白信号传导[15]。β-arrestin 募集是一种普遍存在的 GPCR信号负调控机制,在几乎所有 GPCR 通路中已经得到证实[16],并且该途径是 G 蛋白非依赖性的,可以通过偏倚配体独立地进行药理学调节[17],这可能为 GPCR 信号的功能选择性(偏倚激活)以及 GPCR 药物开发提供了一种新的、通用的、与 G 蛋白无关的方法。这种分析对于筛选 Gαi 偶联的 GPCR 和孤儿 GPCR 特别有利。此外,β-arrestins 不仅可以阻断 G 蛋白信号转导,而且在 GPCRs 的脱敏、内化、复合致敏、细胞增殖反应和基因转录等方面都发挥重要作用[8,18]。
除了上述作用途径,GPCR 可以从细胞内部或在内化过程中的任意时间段内发出传递信号[18-19]。原则上,通过改变配体的物理化学性质,可以改变其在细胞上和细胞内的分布[20]。是否可以通过药物干预来控制内化和再循环/降解的速度还有待研究。
2 GPCRs 药物筛选技术发展历程及功能分析平台
鉴于 GPCR 在正常生理状态和疾病中的重要性,以及它们通过使用小分子作为调节因子进行治疗干预的潜力,GPCR 是目前市场上最大的药物治疗靶点家族,每年的利润在数十亿美元[21]。然而现有的药物筛选技术仍然不同程度地限制了 GPCR 靶点药物的开发,因此,GPCR 相关检测方法和配体的筛选一直处于研究和发展阶段。GPCR 高通量药物筛选技术的发展历程主要包括传统的经典途径和近年来发现的新途径。传统的经典靶向通路涉及机制较单一,测量方法局限于一维层面。最早应用于 GPCR 药物研究的方法是放射性配基结合技术。1970年,Lefkowitz 等[22]使用放射性标记的激素进行了第一次放射性配基结合实验,以确定其受体的结合亲和力。从那时起,3H 或125I 标记的配体开始广泛用于表征化合物对靶 GPCR 的亲和力。然而由于放射性配基标记繁杂、价格昂贵、并且需要复杂的清洗和过滤步骤,导致了其他替代技术的产生,包括依赖于检测G 蛋白介导的第二信使的经典途径,例如 cAMP 检测技术、基于钙流的荧光成像读片器(fluorometric imaging plate reader,FLIPR)技术、基于共振能量转移的福斯特荧光共振能量转移(förster resonance energy transfer,FRET)和生物发光共振能量转移(bioluminescence resonance energy transfer,BRET)技术,以及 G 蛋白非依赖性检测途径——β-arrestin 的募集实验等。此外,对相关报告基因的检测也已广泛使用[23]。这些基于荧光或化学发光检测的功能性分析试剂盒可以从公司购买。
为使筛选方法更可行、筛选结果更准确,GPCR 药物筛选技术的新方法正在逐步更新和开发,并且用于 GPCR的药物筛选工具也更加先进。近年来,出现了多种 GPCR药物开发的新途径,包括荧光素酶双亚基系统、高内涵筛选技术、无标签全细胞分析技术以及虚拟筛选等(表1)。

表1 GPCR 筛选技术总结
2.1 cAMP 检测法
cAMP 由 ATP 生成,是介导神经递质、激素和药物参与多种生理反应的最重要的第二信使之一,也是 GPCR 药物研发途径中的重要一环。如前所述,与 Gαs 和 Gαi/o 蛋白偶联的 GPCR 分别激活或抑制腺苷酸环化酶,从而增加或降低 cAMP 水平。
cAMP 水平通常使用竞争法测量,例如 LANCE cAMP和 HTRF-cAMP 检测试剂盒。LANCE cAMP 试剂盒是一种均相时间分辨荧光能量转移(TR-FRET)免疫测定法。该测定基于铕标记的 cAMP 示踪剂复合物和样品 cAMP 竞争 Alexa Fluor 647 染料标记的 cAMP 特异性抗体上的结合位点。铕标记的 cAMP 示踪剂复合物是由生物素化-cAMP和 Eu-链霉亲和素之间的紧密相互作用形成的。当抗体与Eu-链霉亲和素/生物素-cAMP 示踪剂结合时,340 nm 的光脉冲激发示踪剂的铕螯合物分子,导致能量转移到荧光团,荧光团可在 665 nm 处发光。因此,测试样本中的 cAMP水平越高,竞争水平越高,发出的信号越低,反之亦然(图2)[24-25]。HTRF-cAMP 的检测原理是基于均相时间分辨荧光(homogeneous time-resolved fluorescence,HTRF)技术,该方法是由细胞产生的天然 cAMP 与修饰的别藻蓝蛋白染料标记的 cAMP 之间的竞争性免疫测定。cAMP 检测法可用于高通量药物发现,灵敏度高,但受限于需要清楚偶联机制,不利于筛选 oGPCR 的激动剂或拮抗剂。在cAMP 分析中,筛查 Gs 偶联受体通常较简单,而筛查 Gi/o偶联受体则困难得多。
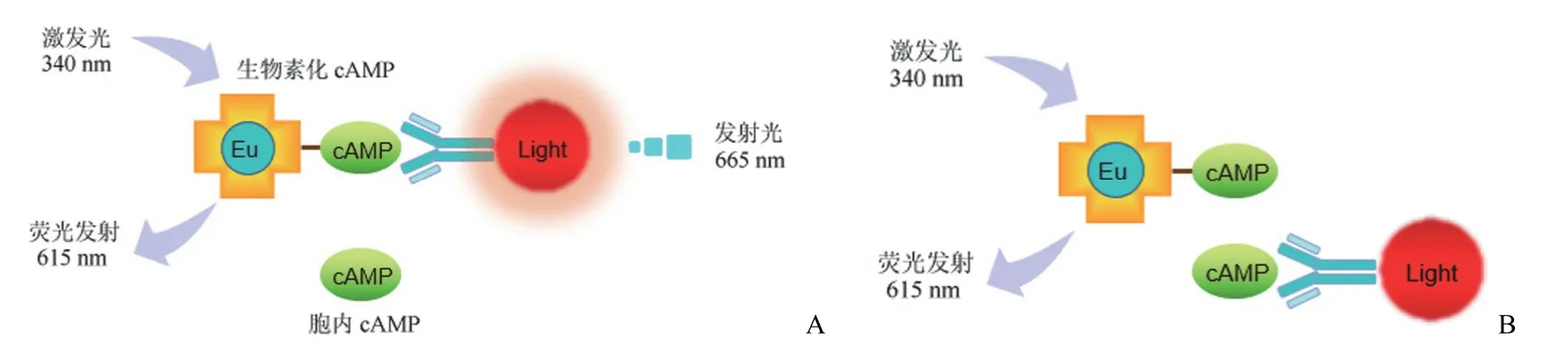
图2 LANCE cAMP 竞争法测定原理[A:在胞内待测 cAMP 不充足的情况下,Eu 标记的 cAMP 探针与抗 cAMP 特异性抗体结合,340 nm 的光脉冲激发示踪剂的铕螯合物分子,导致能量转移到荧光团,荧光团可在 665 nm 处产生荧光信号;B:随着游离 cAMP(细胞 cAMP)浓度的增加,测试样本中的 cAMP 竞争性结合 cAMP 特异性抗体,导致荧光信号减弱]
此外,21 世纪初,BD Biosciences 有限公司开发了ACTOne 检测法,这是一种可以实时检测细胞内 cAMP 水平变化的技术,该分析可以高通量进行,无需细胞裂解步骤,这种检测基于含专有外源性环核苷酸门控(CNG)通道的细胞系,可以对细胞内 cAMP 水平进行终点和动力学测量,并已成功用于测量 Gs 和 Gi 偶联 GPCR 活性[26]。
2.2 基于细胞内 Ca2+ 检测法
在 GPCR 信号通路中,细胞内 Ca2+是 GPCR 信号传导的另一个第二信使。该方法是基于 G 蛋白偶联 GPCR信号通路对细胞内 Ca2+荧光进行定量分析[27]。用于测量细胞中 Ca2+通量的 FLIPR 是 20 世纪 90年代早期的一项重要发明,在高通量药物发现中发挥了重要作用。该法主要用于分析 Gq 偶联的 GPCR,但也可以通过嵌合 G 蛋白或混杂 G 蛋白的表达来检测 Gαi/o、Gαs 或 Gα12 偶联的GPCR[28-30]。
自从 Tsien 及其同事在 1989年开发出荧光钙指示剂fluo-3 以来,基于荧光的细胞内钙测定已变得越来越流行[31],相继开发出了 fluo-3 类似物 fluo-4。Fluo-4 由 Ca2+螯合剂和荧光素类似物组成,一旦与 Ca2+结合,荧光团的电子性质和光谱性质就会发生改变,即可检测到荧光[30]。为增加其细胞膜渗透性,fluo-4 以 fluo-4-AM(乙酰氧基甲基酯)的形式施用于细胞,进入细胞后 fluo-4-AM 被细胞内酯酶水解,导致细胞膜不可渗透性负电荷形式 fluo-45-的产生,fluo-45-与胞内 Ca2+结合从而能够检测到发光信号。然而一些细胞会表达某种有机阴离子转运体,这会导致带负电的 fluo-45-的丢失,从而导致信号降低,这种现象可以通过向细胞中添加丙磺舒(一种转运体抑制剂)来防止(图3)。
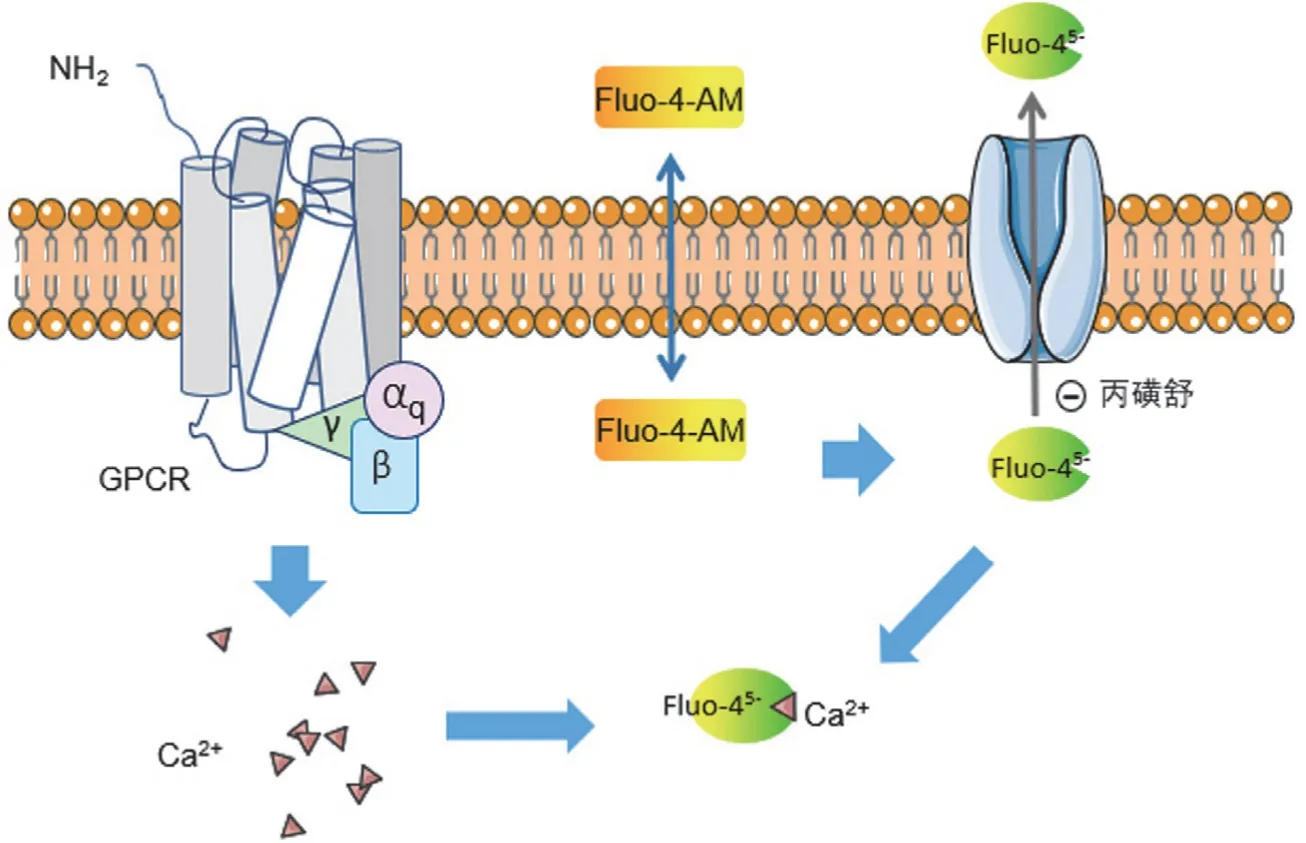
图3 基于荧光钙结合染料 fluo-4 的细胞内钙测定原理
Ca2+测定是活细胞的功能测定,也可用于高通量药物筛选。这种方法可以用于检测激动剂、拮抗剂和变构调节剂,然而由于荧光很容易受到化合物的干扰,因此该法不利于反向激动剂和慢结合激动剂的筛选。
2.3 报告基因分析法
报告基因分析为筛选 GPCR 靶点提供了另一个稳健且经济高效的高通量同质分析平台。GPCR 激活后通过第二信使的响应元件改变基因转录。GPCR 的激活导致 G 蛋白 α 亚基与 βγ 二聚体亚基解离,进而启动下游第二信使通路的级联反应,并最终通过 cAMP 反应元件(CRE)、活化 T 细胞核因子响应元件(NFAT-RE)、血清响应元件(SRE)和血清反应因子响应元件(SRF-RE)等各种反应元件诱导基因转录[32-33](图1)。常用的报告基因包括荧光素酶、碱性磷酸酶、β-半乳糖苷酶、β-内酰胺酶和多种荧光蛋白。荧光素酶通常是首选的报告基因。例如,通过使用位于荧光素酶基因上游的 T 细胞反应元件(NFAT-RE)监测与 Gq 蛋白偶联的受体的活性。受体的激活导致细胞内Ca2+的增加,进而激活蛋白激酶 C 使 NFAT-RE 结合蛋白磷酸化。当磷酸化的 NFAT-RE 结合蛋白与 NFAT-RE 序列结合时,细胞内的荧光素酶浓度会增加,从而导致荧光素酶基因的转录率增加。该平台属于均相测定,需要较长的孵育时间,并且测量的信号事件远离受体激活时间,这可能会导致大量假阳性,并且该法受限于需要了解耦合机制,因此不利于对孤儿 GPCR 的筛查。
2.4 共振能量转移检测法
共振能量转移(resonance energy transfer,RET)是一种光物理过程,通过荧光供体将能量转移到合适的荧光能量受体而检测发光信号,它们在描述活细胞中的 GPCR 激活和信号通路方面非常有价值。根据标签的不同,RET 传感器可分为FRET 和 BRET[34-35]。FRET 的原理是荧光共振能量转移,当供体和受体彼此非常接近时,光子可以从一个受激发的荧光团(供体)转移到另一个荧光团(受体)并使后者发光[36]。BRET 通常是基于发光酶——荧光素酶进行生物发光共振能量转移(图4)[37]。例如,G 蛋白 α(能量供体)和 γ(能量受体)亚单位可以用相应的荧光蛋白标记,GPCR 激活后即可获得 BRET 信号[38-39]。BRET 已被用于研究融合到肾素荧光素酶(Rlu)的 GPCR 与融合到GFP 的细胞质支架蛋白 β-arrestin 的相互作用,以及识别GPCR 的偏倚效应、变构和多谱关联[40]。
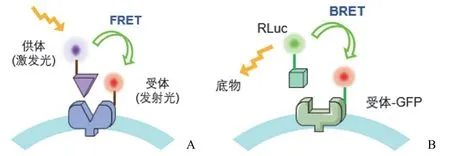
图4 FRET 和 BRET 检测两种蛋白质之间的能量转移[A:FRET 中有两个荧光团,当供体和受体彼此靠近时,激光激发能量供体荧光团,光子从该供体转移到另一个荧光团(受体)并使后者发光;B:BRET 中当两个荧光团靠近时,荧光素酶(供体)与底物反应发出的荧光可以激发受体上的 GFP 荧光团,GFP 随后发出更高波长的可检测信号。其中 FRET 需要外部激光激发,而 BRET 是采用生物发光]
根据实验的具体情况,RET 传感器可以根据标记单位的不同分为几种表达方式,如 GPCR/G 蛋白信号功能测定、GPCR/β-arrestin 信号功能测定[41]、分子内构象 GPCR传感器和分子内构象 GPCR 二聚化传感器[42]。FRET 和BRET 的实际应用取决于具体的实验情况,FRET 测定提供了更高的空间和时间分辨率,荧光探针的高度多样性提供了广泛的探针对,但是这种技术需要外部光源来激发供体,并且高背景会导致信噪比降低。相比之下,BRET 检测是基于酶促反应,不需要额外的激发光,更容易量化结果[36]。
2.5 G 蛋白非依赖的 β-arrestin 招募法
基于 β-arrestin 的分析方法是一种不依赖于 G 蛋白的测定方法,特别适用于 oGPCR 的分析和筛选。研究发现,有些 GPCRs 的配体在激活信号通路时呈现出一种“不平衡效应”,它们能够诱导受体选择性地偶联不同类型的 G 蛋白亚基或与非 G 蛋白调控因子 β-arrestin 直接作用[43-45],从而使胞内的信号偏向某一通路,这种配体称为“偏向性配体”或“偏倚配体”[17]。因此针对一种 G 蛋白的特殊信号通路的功能性药物筛选分析可能会导致对这种偏倚配体的忽略。此外,通过异源二聚体的受体串扰可使受体信号通路更加复杂[46]。
最近开发了多种基于 β-arrestin 招募的药物筛选分析方法,可以通过联合 BRET、PathHunterTM技术和 TangoTM技术进行检测。BRET 是最早用于监测 GPCR-β-arrestin 相互作用的分析方法之一[47],该技术的分析原理是将 GPCR的 C 端和 β-arrestin 分别用海肾荧光素酶(RLuc)和荧光蛋白标签(eGFP2、GFP10 或 YFP)标记[48],当 β-arrestin被招募时,这两个标签彼此非常接近,RLuc 反应发出的光激发荧光蛋白,然后荧光蛋白发出更高波长的用于信号检测(图5A)。BRET 已应用于包括趋化因子、阿片类、多巴胺和前列腺素受体等在内的 GPCR 靶向药物筛选[49-50]。在PathHunterTM检测中,β-arrestin 与无催化活性的 β-半乳糖苷酶缺失突变体融合,在 GPCR 的 C 端用 β-半乳糖苷酶N 端缺失序列的另一个小片段标记,GPCR-β-arrestin 相互作用后,β-半乳糖苷酶的两个部分紧密靠近而活化,导致底物裂解并产生化学发光信号(图5B)[51]。
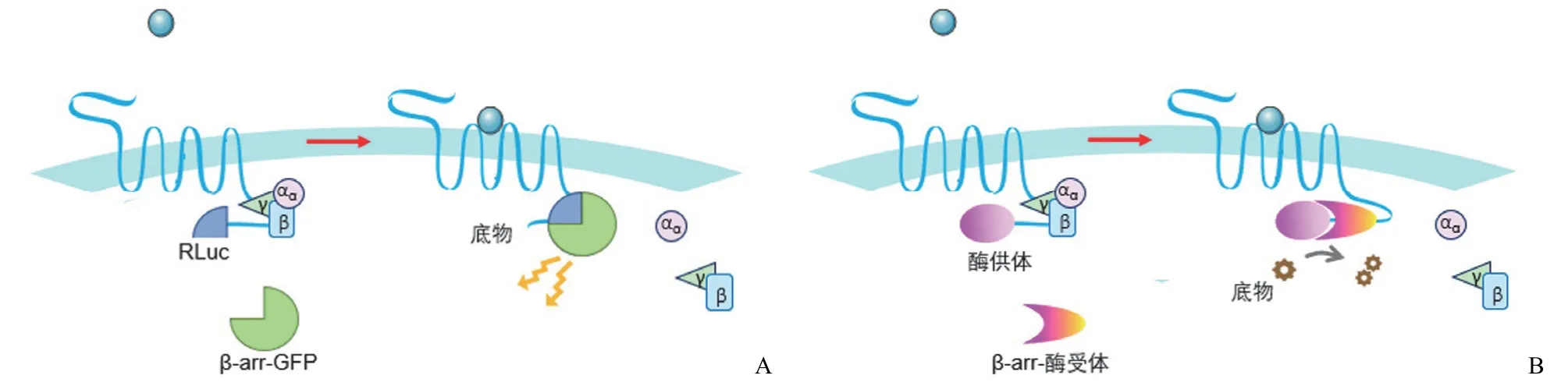
图5 β-arrestin 招募分析(A:BRET 实验;B:PathHunterTM 实验)
2.6 NanoBiT 荧光素酶检测系统
最近,荧光素酶双亚基系统新技术开发成功。NanoLuc二元技术(NanoBiT)包括一个 18 kD 的大亚基片段 LgBiT和一个 1.3 kD 的小亚基片段 SmBiT,该系统可用于细胞内蛋白质相互作用的检测。LgBiT 和 SmBiT 亚基分别与靶蛋白融合[52],这两个亚基之间的内在亲和力极低(Kd= 190 μmol/L),当目的蛋白表达时,蛋白-蛋白相互作用(protein-protein interactions,PPI)使两个亚基靠近而形成一种功能性酶,进而能够催化底物产生明亮的发光信号[53-54]。在 NanoBiT 系统的开发中,鉴定出另一种不同于SmBiT 的小互补肽-HiBiT,它是一个仅有 11 个氨基酸的序列,对 LgBiT 具有非常高的亲和力(Kd= 700 pmol/L)。由于 LgBiT 是细胞不可渗透的,因此 HiBiT-LgBiT 互补系统提供了一种区分内化蛋白质和保留在细胞表面的蛋白质的方法(图6)。
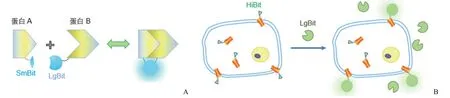
图6 NanoBiT 荧光素酶检测系统[A:蛋白质-蛋白质相互作用检测(将目的蛋白 A 和 B 分别与 LgBiT 和 SmBiT 融合,并在细胞内表达。融合蛋白的相互作用导致 LgBiT 与 SmBiT 的结构互补,形成可产生明亮发光信号的有功能的酶);B:Nano-Glo HiBiT 细胞外检测系统(将目的蛋白标记 HiBiT 标签并在细胞内表达,当外源性加入 LgBiT 蛋白和底物后,两者结合形成复合物产生明亮的发光酶,通过检测荧光强度,可判断细胞膜表面或分泌到胞质的 HiBiT 标记的目的蛋白量)]
NLuc 系统用途广泛,已用于生物医学研究的若干领域,包括蛋白质-蛋白质相互作用、遗传调节和细胞信号、监测蛋白质稳定性和分子成像等。据报道,NLuc 已经成功地监测了 G 蛋白偶联松弛素家族肽受体(RXFP3)的内化,通过在 HEK 293T 细胞中表达 NLuc 标记的 RXFP3,利用生物发光来量化检测结果[55]。此外,已有研究通过基因组编辑技术将 HiBiT 标签标记到程序性死亡配体 1(PD-L1)的 C 端,并在肺腺癌细胞系 PC9-KI 中,用药物筛选鉴定调节 PD-L1 表达的化合物[56]。
NLuc 技术与其他系统相比具有多种优点。首先,它将成为更准确的 PPI 生物学模型。LgBiT 和SmBiT 都是较小的蛋白标记物,对目标蛋白的构象干扰极微弱。第二,NanoBiT 具有更高的灵敏度和信噪比[57-58]。第三,由于LgBiT 和 SmBiT 之间的相互作用是可逆的,该系统还可用于检测结合后快速分离的蛋白质。第四,NanoBiT 检测方法简单,无需特定过滤器或进样器,用普通发光检测器即可检测,并且试剂稳定性高。第五,该法可调节放大测试通量,适用于 96 孔、384 孔和 1536 孔板的高通量检测,以用于药物开发[59-60]。此外,该方法不受特定 G 蛋白类型的限制,可用于筛选 Gαs 和 Gαi/o 以及任何其他相偶联的 G 蛋白亚家族。
2.7 其他新兴药物筛选技术
近年来,通过高分辨率荧光显微镜和自动图像分析使GPCR 可视化的高内涵筛选(high-content screening,HCS)成为另一种重要的药物筛选分析方法[61-62]。HCS 主要用于细胞毒性、受体调节剂和活性物质释放等影响细胞功能方面的研究。在药物筛选方面,HCS 也发挥了重要作用。与传统的细胞分析法相比,HCS 可以识别每个细胞中的个体反应,从而获得细胞周期阶段、细胞转染率或自然变异性的可靠数据。目前,许多制药和生物技术公司已将 TransFluor 检测法确认为 HCS 金标准分析。据报道,Ross 等[63]已利用TransFluor 技术通过检测荧光信号强弱与分布及其变化对GPCR 进行高内涵筛选。
无标签技术极大地推进了 GPCR 全细胞分析手段,该方法使用生物传感器,将配体诱导的活细胞变化总和转换为光学、电学、量热、声学、磁性或其他可量化信号,可以用于检测包括细胞黏附、增殖、迁移和死亡的特征性变化。目前基于光学的仪器包括 BindTM(SRU 生物系统)和 EpicTM(康宁公司)系统,这两个系统都已成功地用于研究细胞形态变化和 GPCR 信号通路[64]。但无标签全细胞分析也容易出现假阳性和假阴性结果[65]。
基于结构的药物设计长期以来在药物发现中发挥着重要作用,尤其是对于具有酶靶点的药物。目前,基于 GPCR结晶学的最新突破[66-67],发现 44 种不同的 GPCR 结构和196 种配体-受体复合物可用于人类 GPCR 研究,这为虚拟筛选和细胞非依赖发现新药物提供了重要依据[68-69]。例如,最近针对 μ 阿片受体结构的对接实验确定了一种 Gi 蛋白偏向的激动剂 PZM21,其疗效与吗啡相似,但减少了对小鼠的不良反应[70]。与传统分析方法相结合,这些新兴的药物筛选平台在 GPCR 药物筛选和开发中有着广阔的发展前景。
3 小结
GPCR 靶向药物是学术界和制药界最广泛的研究焦点。GPCR 蛋白家族在糖尿病、肥胖、阿尔茨海默症等为代表的疾病中具有重要临床用药价值。此外,一些孤儿受体已锁定为多种适应证的新靶点,这为该领域的药物研发工作提供了强大动力。由于 X 射线晶体学、蛋白质工程和生物物理技术的发展,GPCR 先导化合物的开发技术正在逐渐迭代更新,新的分析方法更加高效和多维,例如 3D 晶体学结构分析、无标记荧光分析和虚拟筛选等。
尽管如此,让 GPCR 靶向药物真正服务于临床仍然是一项具有挑战性的工作,这对高通量筛选技术的工艺优化和新技术开发提出了更高的要求。展望未来,基于结构和传感器的方法与传统的 HTS 相结合,可能会为 GPCR 药物发现领域带来新的机遇,从而扩大潜在靶向界面范围,为更精准的药物治疗提供可能性。
猜你喜欢
保健医苑(2022年5期)2022-06-10
中国临床医学影像杂志(2021年6期)2021-08-14
肝博士(2020年5期)2021-01-18
心肺血管病杂志(2019年1期)2019-04-22
材料科学与工程学报(2016年4期)2017-01-15
当代化工研究(2016年9期)2016-03-20
合成化学(2015年4期)2016-01-17
中国塑料(2015年8期)2015-10-14
医学研究杂志(2015年7期)2015-06-22
无机化学学报(2014年6期)2014-02-28
