
纤维微菌β-1,3-葡聚糖酶的克隆表达及对灵芝细胞壁的水解作用
2023-02-03 07:05杨梦莲单逸蓝沈微朱新文杨海泉陈献忠陈磊夏媛媛
食品与发酵工业 2023年1期
杨梦莲,单逸蓝,沈微*,朱新文,杨海泉,陈献忠,陈磊,夏媛媛
1(江南大学 生物工程学院 工业生物技术教育部重点实验室,江苏 无锡,214122)2(山东省东阿县农业农村局,山东 聊城,252200)
硒是人体必须的微量元素之一,是人体多种含硒蛋白的组成成分[1]。硒元素也被证实具有抗氧化、提高机体免疫力、养肝护肝、预防心脑血管疾病等生理功能[2-3]。硒元素在地球上分布不均匀。我国地域辽阔,环境多样,富硒和缺硒地区并存,但全国范围来讲,我国是缺硒范围广、程度严重的国家之一[4]。无机硒元素被生物体吸收后形成有机硒,有机硒更容易被人体吸收,具有更高的安全性[3,5]。
灵芝是我国医药和食疗宝库中的珍品,以灵芝为富硒载体,将无机硒转化为有机硒,获得的富硒灵芝有可能实现硒和灵芝固有生理作用的结合[6]。国内外科研工作者对灵芝富硒进行了大量的研究[7-9]。富硒灵芝不单是硒元素的补充剂,部分含硒生物分子还具有特殊的生理功能[9]。一些灵芝硒多糖具有明显的抗肿瘤或诱导癌细胞凋亡的作用[10-12],而一些含硒多肽则具有较强的抗氧化功能[1,13]。
富硒灵芝中的有机硒以蛋白硒、多糖硒、核酸硒等多种形式存在,其中蛋白硒是有机硒分子的主要存在形式[14]。蛋白质和核酸大多存在于灵芝的细胞内,也有一部分多糖存在于细胞内[14-15]。因此为获取目标产物需对灵芝细胞壁进行破碎。超声波破碎法原则上适用于各种生物细胞的破碎,但用超声波方法破碎大型真菌的菌丝体细胞一般需要相当长的时间而且蛋白得率较低,在工业化生产时有很大的困难。在各种细胞破壁方法中,酶解法具有条件温和、操作方便、提取效率高等优点[16]。
到目前为止,有关灵芝菌丝体细胞壁酶解的研究报道较少。用于灵芝菌丝体破壁的酶有酵母溶壁酶、蜗牛酶等,这些酶处理灵芝菌丝体可以获得一定量的原生质体,但这些酶目前仅作为科研试剂使用,价格昂贵,不具备工业化应用的条件。本文作者在前期工作中对实验室保藏的微生物进行筛选,发现1株纤维微菌(Cellulosimicrobiumsp.)的发酵液与少量灵芝菌丝体混合后可以获得原生质体。进一步发现,这株菌的β-1,3-葡聚糖酶基因的表达产物与市售果胶酶联合处理灵芝菌丝体可以获得原生质体,进一步辅以短时超声波处理可以有效实现灵芝菌丝体细胞的破碎。本文报道该基因的克隆表达及其与果胶酶联合使用提取灵芝胞内蛋白的方法。
1 材料与方法
1.1 实验材料
1.1.1 菌株和质粒
表达宿主菌大肠杆菌EscherichiacoliBL21(DE3)、表达载体pET-28a(+)为本课题组保藏;纤维微菌(Cellulosimicrobiumsp.)CICIM B6906,中国高校工业微生物资源平台,简称纤维微菌6906;灵芝(Ganodermalucidum)ML2613已分别保藏在中国高校工业微生物资源平台(CICIM F7083)和中国典型培养物保藏中心(CCTCC No:M 20211217)。
1.1.2 主要试剂
限制性内切酶、连接酶,赛默飞世尔科技公司;质粒提取和DNA片段回收试剂盒,苏州康宁生物科技有限公司;DL10000 DNA Marker,宝生物(大连)生物工程公司;蛋白标准分子量,翌圣生物科技有限公司;茯苓多糖、酵母葡聚糖、大麦葡聚糖、羧甲基纤维素(carboxymethylcellulose, CMC)热凝胶,MegaZyme公司;热凝胶、昆布多糖、微晶纤维素等,上海源叶生物技术公司;果胶酶等酶制剂,南宁庞博生物工程有限公司;His-Trap HP 1 mL亲和层析柱,美国GE公司;其余试剂均购自国药集团化学试剂有限公司。
1.1.3 主要培养基的配制
LB和TB培养基配方按文献[17]的方法配制。
重组大肠杆菌分批补料发酵基础培养基(g/L):酵母粉30,蛋白胨15,甘油10 mL/L,KH2PO42.31,K2HPO4·H2O 16.43,(NH4)2SO42,MgSO4·7H2O 0.2,硫胺素0.02。121 ℃,实罐灭菌15 min。灭菌后1 L培养基中加入微量元素溶液0.05 mL。
微量元素溶液(g/L):FeSO4·7H2O 2,ZnSO4·7H2O 0.4,CuSO4·5H2O 0.2,MnSO4·4H2O 0.1,Na2B4O7·10H2O 0.04,(NH4)6Mo7O240.02。溶解后用无菌过滤器过滤除菌。
补料培养基(g/L):甘油500 mL/L,MgSO4·7H2O 0.3,酵母粉50,蛋白胨50。
诱导培养基(g/L):乳糖200。
灵芝种子培养基(g/L):葡萄糖20,麦芽汁粉20。
灵芝摇瓶发酵培养基(g/L):葡萄糖30,KH2PO40.2,尿素0.1,麦芽汁粉20,酵母粉5。发酵至24 h后加入Na2SeO3·5H2O至180 mg/L。
灵芝发酵罐发酵培养基(g/L):葡萄糖40,KH2PO40.2,(NH4)2SO42,CaCl20.1,MgCl20.05,麦芽汁粉10,酵母粉5。115 ℃,灭菌15 min。发酵24 h后加入Na2SeO3·5H2O至180 mg/L。发酵过程用100 g/L NaOH溶液控制pH 4.5~5.0。
1.1.4 引物
根据菌种鉴定结果,纤维微菌6906的16S rDNA基因序列与NCBI公布的纤维微菌JZ28的最为接近,而JZ28的基因组序列(NCBI登录号:CP017660.1)中有一段与β-1,3-葡聚糖酶有较高相似性的序列(上述基因组序列中231 041~232 687碱基之间的片段),根据该序列设计引物如下:
Pgl101:5′-aatcggatcctaaatacttaaggaggattcttatgcctcacgacagg-3′
Pgl102:5′-aatcaagcttgagcgtccagcgct-3′
Pgl101带单下划线部分为翻译终止密码子,用于终止载体pET-28a(+)多克隆位点上游开始的包含His-tag在内的短肽的翻译。双下划线部分为SD序列和翻译起始密码子,从atg往后14个碱基与所预测的葡聚糖酶基因序列一致,以上结构控制以载体T7启动子起始转录的mRNA从葡聚糖酶基因的ATG开始独立翻译。引物Pgl102与所预测葡聚糖酶基因的3′端互补,但删去了翻译终止密码子tag,由此控制所插入葡聚糖酶基因的翻译能延续到载体多克隆位点下游的His-tag标签的编码区,便于表达产物的纯化。2条引物分别带有内切酶BamH I、Hind III识别位点。
1.2 实验方法
1.2.1 β-1,3-葡聚糖酶重组表达载体的构建
提取纤维微菌6906基因组DNA,以基因组DNA为模板,以Pgl101、Pgl102为引物进行PCR扩增,PCR产物用BamH I和Hind III酶切,纯化后与经同样酶切的载体pET-28a(+)连接,转化大肠杆菌JM109,涂布含50 μg/mL卡那霉素的LB平板,任取几个转化子进行液体培养,提取转化子质粒进行酶切验证和测序。验证正确的转化子转化表达宿主菌大肠杆菌BL21(DE3)。
1.2.2 重组β-1,3-葡聚糖酶的表达及产物的分析
将重组菌株在卡那霉素50 μg/mL的LB固体培养基上划线,挑取单菌落接种至含卡那霉素50 μg/mL的LB液体培养基,于37 ℃、200 r/min过夜培养后,按1%(体积分数)接种量接入到50 μg/mL卡那霉素的TB液体培养基中,于37 ℃、200 r/min培养至菌体密度OD600达到0.6左右,加入终浓度为0.5 mmol/L的IPTG,于30 ℃、200 r/min诱导培养24 h。发酵结束后,发酵液7 000 r/min离心10 min。其中上清液作为胞外粗酶液直接检测酶活力。沉淀部分加入与发酵液等体积的磷酸缓冲液悬浮菌体后超声波破碎检测酶活力,方法同文献[17]。胞外粗酶液蛋白电泳前先用离心式超滤浓缩管(30KD/UFC900396,Millipore)按产品说明书进行浓缩,一般是浓缩至1/10体积。
1.2.3 重组β-1,3-葡聚糖酶的纯化及酶活力测定
重组酶的C-端带有His-tag标签,所以采用镍柱进行亲和层析纯化,具体纯化方法如下:取胞外粗酶液5 mL,经0.22 μm滤膜过滤,用含有0.5 mol/L NaCl和0.02 mol/L咪唑的Na2HPO4- NaH2PO4缓冲液(0.02 mol/L,pH 7.4)作为上样缓冲液,用含0.5 mol/L咪唑和0.5 mol/L NaCl的Na2HPO4- NaH2PO4缓冲液作为洗脱液,梯度洗脱,流速为1 mL/min。SDS-PAGE分析纯化结果,用考马斯亮蓝法测定蛋白质含量,计算比酶活力,方法同文献[17]。
采用DNS法测定β-1,3-葡聚糖酶的活力。取50 μL适当稀释的酶液,加入100 μL 15 g/L的茯苓多糖(由0.1 mol/L柠檬酸与0.2 mol/L Na2HPO4构成的缓冲液配制,pH 5.5),用缓冲液将其定容到500 μL,50 ℃下反应15 min,加入750 μL DNS试剂,沸水浴煮10 min,在540 nm下测定吸光度。根据葡萄糖标准曲线计算还原糖生成量。酶活力定义:将每分钟水解茯苓多糖产生的还原糖,其还原力相当于1 μmol葡萄糖所需的酶量定义为β-1,3-葡聚糖酶的1个酶活力单位(U)。
1.2.4 重组β-1,3-葡聚糖酶酶学性质的测定
重组酶最适温度的测定:分别在30~65 ℃内间隔5 ℃测定酶活力,以最高酶活力为100%,计算相对酶活力。
重组酶最适pH的测定:在pH为3.5~8.5的Na2HPO4-柠檬酸缓冲液中,测定酶活力,以最高酶活力为100%,计算相对酶活力。
重组酶热稳定性的测定:将酶液置于40、45、50、55 ℃金属浴中保温3 h,分别测定保温0.5、1、1.5、2、2.5、3 h后的酶活力,以未处理的酶活力为100%,计算相对酶活力。
1.2.5 重组酶对不同底物特异性研究
分别以15 g/L的大麦葡聚糖、茯苓多糖、热凝胶、酵母葡聚糖、昆布多糖、地衣多糖和微晶纤维素等为底物,在最适条件下测定重组酶的活力,分析重组酶对不同底物的水解能力。
1.2.6 重组β-1,3-葡聚糖酶的发酵
取重组菌单菌落接种于含50 μg/mL卡那霉素的LB培养基中,摇瓶培养过夜,转接于新鲜的含卡那霉素的LB培养基培养至OD600≈1,用作种子培养液。15 L发酵罐中预先加入10 L基础发酵培养基,并实罐灭菌。灭菌后降温至37 ℃,控制通气量12 L/min,发酵罐搅拌转速控制在250 r/min,校正溶氧量为100%,适当补充体积分数25%氨水控制发酵pH在6.5~7.0。以2%(体积分数)的接种量接入种子培养液,同时加入终质量浓度为50 μg/mL的卡那霉素,发酵过程偶联转速使溶氧维持在20%~30%。培养至发酵罐中的溶氧浓度持续上升时(即甘油耗尽时),开始以25 mL/h的速度流加补料培养基,并继续偶联转速使溶氧维持在20%~30%,始终以25%氨水控制发酵pH在6.5~7.0。发酵至32 h,发酵液OD600约为100时加入乳糖至终质量浓度为5 g/L进行诱导表达。当发酵液中的胞外酶活力不再明显增加时,结束发酵。发酵液离心后取上清液进行超滤浓缩。使用UF101小型超滤机,超滤膜为PES30-1512,截留分子质量30 kDa(上海福立特实业有限公司产品),超滤压力控制在(0.1±0.01) MPa。
1.2.7 灵芝菌丝体发酵与菌体的裂解
将灵芝ML2613的菌丝体斜面培养物接种于灵芝种子培养基中,在30 ℃、180 r/min的摇床中培养3 d,按10%(体积分数)接种于5 L发酵罐的发酵培养基中。发酵培养基装液量为3 L,发酵24 h后加入180 mg/L的Na2SeO3·5H2O,培养5 d,过程中用50 g/L的NaOH调节pH在4.5~5.0。将培养后的菌丝体进行过滤,用于菌体破碎实验。
灵芝菌丝体的酶解方法如下:取适量灵芝菌丝体,用缓冲液悬浮后加入酶液至适当浓度,在40 ℃反应。对照实验中,首先将所使用的酶液在90 ℃保温20 min,将酶灭活,再按上述方法同样进行菌丝体酶解。取等量的用酶处理过的灵芝菌丝体和按对照方法处理的灵芝菌丝体分别进行超声波破碎,超声波功率为300 W。超声波破碎后采用碱提法提取裂解液蛋白质。用1.0 mol/L NaOH溶液将料液pH调至10.0,浸提1 h,以促进蛋白质在碱性条件下充分溶解,最后将料液7 000 r/min离心10 min,上清液为灵芝提取液。采用考马斯亮蓝法测定提取液中蛋白质含量[17]。蛋白提取率的计算如公式(1)所示:

(1)
未经处理的灵芝菌丝体按GB 5009.5—2016《食品中蛋白质的测定》,采用凯氏定氮法测定灵芝菌丝体总蛋白。
摇瓶实验时以新华滤纸为介质采用抽滤方法过滤收集菌体。发酵罐实验时采用150型小型板框压滤机(嘉兴优创机械设备有限公司)过滤收集菌体,滤布为510型(孔径5~10 mm)丙纶滤布。
1.2.8 灵芝蛋白含硒量检测
上述得到的提取液用HCl溶液调节至等电点使其沉淀,后续采用(NH4)2SO4盐析沉淀进一步纯化得到灵芝蛋白粉,方法同文献[18]。蛋白粉按GB 5009.268—2016《食品中多元素的测定》,采用电感耦合等离子体质谱法(inductively coupled plasma mass spectrometry, ICP-MS)测定富硒灵芝粗蛋白中的硒含量。
2 结果与分析
2.1 重组菌的构建
以纤维微菌6906的染色体DNA为模板,以Pgl101、Pgl102为引物进行PCR扩增,扩增产物为1.7 kb左右,与推测的纤维微菌β-1,3-葡聚糖酶基因大小一致,该基因片段命名为bgl6906。以核酸内切酶BamH I和Hind III酶切上述目的片段,与经同样酶切的表达载体pET-28a(+)连接后转化大肠杆菌JM109,挑取转化子提取质粒,酶切验证。结果表明,正确转化子所含质粒酶切后获得5.2和1.7 kb的2个片段,与pET-28a(+)空质粒和扩增片段bgl6906一致,图1是正确转化子质粒酶切电泳图,该重组质粒命名为pET28a-bgl6906。

M:DL10000 DNA marker;1:pET28a-bgl6906图1 重组质粒pET28a-bgl6906酶切图谱Fig.1 Restriction analysis of the recombinant plasmid pET28a-bgl6906
重组质粒测序并将序列NCBI进行序列搜索比对,结果显示该序列的编码区与NCBI核苷酸序列库中收录的纤维微菌Cellulosimicrobiumsp.JZ28基因组序列中的一段尚未标注的序列最为接近,两者翻译成氨基酸序列后同源性为98%。基因bgl6906编码区全长1 644 bp,编码1条含有548个氨基酸残基,理论分子质量为58 kDa的多肽链。用在线分析软件SignalP(https://services.healthtech.dtu.dk/)分析显示,上述多肽链N端有1段36个氨基酸残基组成的信号肽,剩余的512氨基酸残基的成熟肽分子质量为54.4 kDa(不含6×His部分)。对Blast比对结果进行分析显示,bgl6906基因编码的蛋白序列与藤黄节杆菌(又名纤维素纤维微菌)的β-1,3-葡聚糖酶氨基酸序列[19]同源性为96%。上述藤黄节杆菌也是比对结果中,有表达产物酶学性质报道的序列中与本文所克隆bgl6906最接近的基因。
2.2 重组β-1,3-葡聚糖酶的分泌表达及纯化
将重组质粒pET28a-bgl6906转化表达宿主菌大肠杆菌BL21(DE3),转化子命名为大肠杆菌BL21(DE3)/pET28a-bgl6906,简写为BL21/6906。将重组菌BL21/6906以及对照菌即pET-28a(+)空质粒的转化子分别按1.2.2的方法表达制备粗酶液并检测酶活力。结果显示,对照菌细胞内外均没有酶活力。BL21/6906的细胞内外均有明显酶活力,其中发酵液上清液部分的酶活力大约为11 U/mL,而细胞破碎液酶活力大约为3 U/mL。从实验结果看,bgl6906基因所编码信号肽在大肠杆菌中也同样具有引导重组蛋白向细胞外分泌的功能。由于bgl6906基因所编码蛋白与藤黄节杆菌来源的β-1,3-葡聚糖酶的氨基酸序列有明显的差异,两者的酶学性质不一定完全相同,因此我们将上述转化子的表达产物纯化并对酶学性质进行了分析。
蛋白纯化结果如图2所示。对比1、2泳道可见,与对照菌相比,BL21/6906在分子质量55 kDa附近有一微弱的差异表达条带,利用His标签纯化后在泳道3获得纯酶的单一条带,泳道3纯酶的条带位置与上述1、2泳道的差异表达条带分子质量一致。测定纯酶酶液的酶活力及蛋白浓度,经过计算以茯苓多糖为底物时该重组蛋白的比酶活力为567.8 U/mg。为便于叙述,重组菌BL21/6906表达的重组酶命名为BGL6906。

M-蛋白标准分子量;1-BL21(DE3)/pET28a(+)胞外组分10倍浓缩液;2-BL21/6906胞外组分10倍浓缩液;3-BGL6906纯酶图2 重组菌株胞外蛋白SDS-PAGE分析Fig.2 SDS-PAGE analysis of extracellular protein of the recombinants
2.3 重组酶BGL6906的酶学性质
2.3.1 重组酶BGL6906的最适温度和最适pH
在pH 5.5的缓冲液中,30~75 ℃测定BGL6906的相对酶活力,结果如图3所示。BGL6906在35~50 ℃之间随着温度的提高,酶活力逐步提高,50 ℃为最适温度,在45~55 ℃内BGL6906的酶活力均在最高酶活力的80%以上。温度超过60 ℃时酶活力迅速下降。

图3 温度对BGL6906酶活力的影响Fig.3 Effects of temperature on the activity of BGL6906
在50 ℃下,测定不同pH值的酶活力,结果如图4所示。
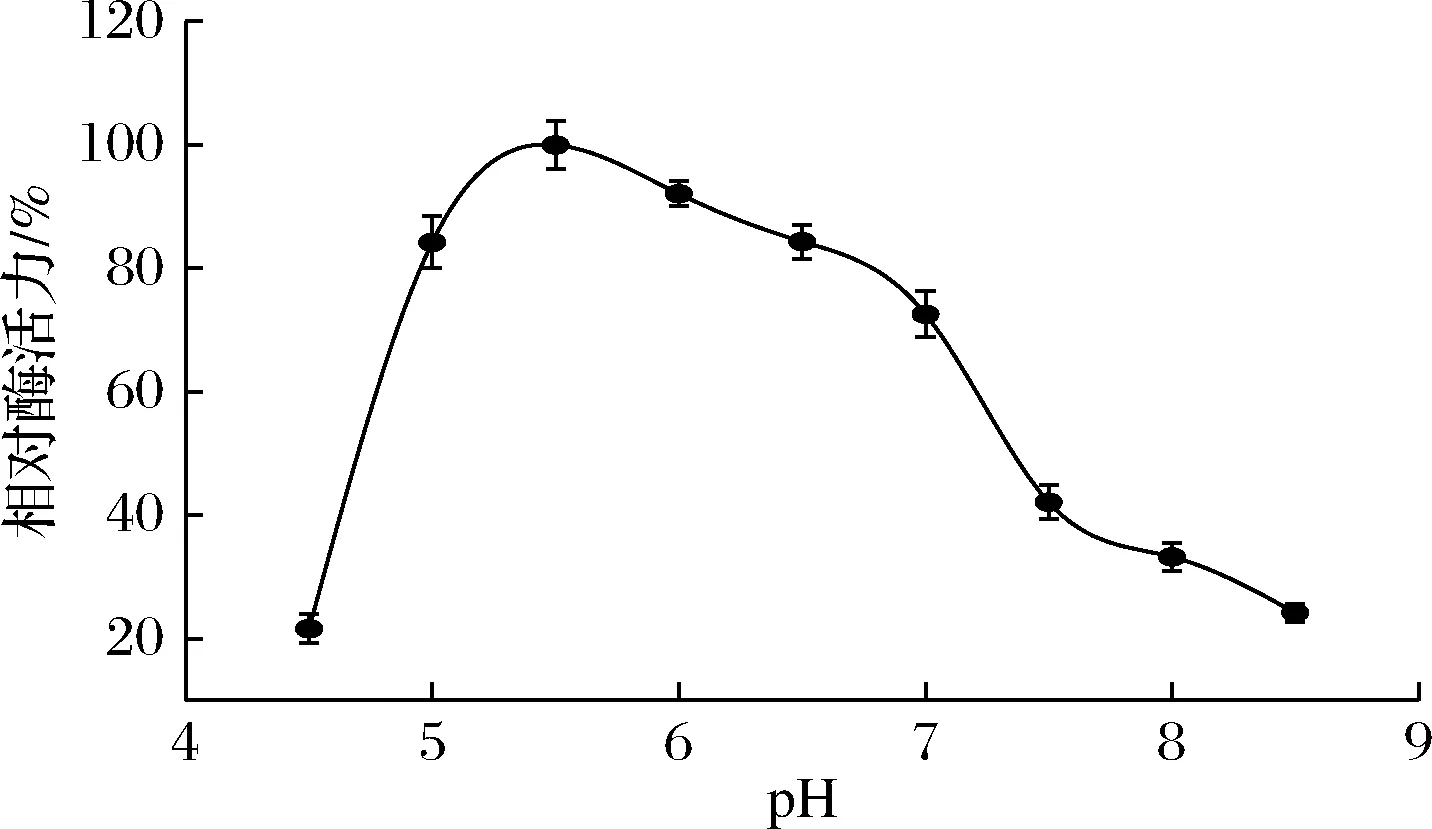
图4 pH对重组酶BGL6906酶活力的影响Fig.4 Effects of pH on the activity of the recombinant BGL6906
在pH 5.0~6.0内,BGL6906酶活力均在最高酶活力的80%以上,5.5为最适pH值。pH低于5.0和超过7.0时,酶活力明显下降。
2.3.2 重组酶BGL6906的热稳定性
将酶液置于45、50、55、60 ℃金属浴中处理3 h,间隔0.5 h测定相对残余酶活力,结果如图5所示,45 ℃保温处理3 h,残余酶活力在70%以上;50 ℃保温处理3 h,残余酶活力在55%以上。温度超过50 ℃,酶活力下降迅速,55 ℃保温处理3 h,相对酶活力仅为11%。在60 ℃条件下,BGL6906在30 min后只剩下大约40%的酶活力,1.5 h后完全失活。

图5 重组BGL6906的热稳定性Fig.5 Thermo-stability of the recombinant BGL6906
2.3.3 重组BGL6906的底物特异性
将重组酶分别与15 g/L的不同底物反应,研究其对于不同底物的水解能力。结果如表1所示,BGL6906水解茯苓多糖的活力最高,比酶活力达到567.8 U/mg。BGL6906水解酵母葡聚糖、热凝胶和昆布多糖的比活力约为86.2、23.04和16.1 U/mg。大麦葡聚糖、地衣多糖、CMC-热凝胶为底物时,BGL6906均未表现出明显的酶活力。BGL6906的底物特异性与纤维微菌属来源的其他β-1,3-葡聚糖酶表现出相似的底物特异性[20],即只对以β-1,3-糖苷键为主的葡聚糖表现出明显的水解活性。

表1 重组酶BGL6906的底物特异性Table 1 Substrate specificity of recombinant BGL6906
2.4 重组菌BL21/6906发酵罐发酵结果
按照1.2.6的补料发酵工艺进行15 L发酵罐发酵,结果如图6所示。在发酵至32 h前,发酵液几乎没有酶活力,32 h时由于添加了诱导剂乳糖酶活力开始快速增长,但同时菌体增长速度快速下降。从蛋白电泳的情况看,胞外重组酶只占胞外蛋白的极少部分,因此基因表达对细胞营养物质的消耗并不大,菌体生长快速下降的原因可能是重组酶对重组菌具有一些尚不明确的不利影响。在补料分批发酵条件下,重组菌最终胞外酶活力为67 U/mL左右,体积大约为11.5 L。为便于后续细胞裂解实验,将发酵液离心去除细胞后用超滤机对酶液进行浓缩,单罐发酵液平均可获得浓缩酶液2.0~2.2 L,酶活力约为320 U/mL。
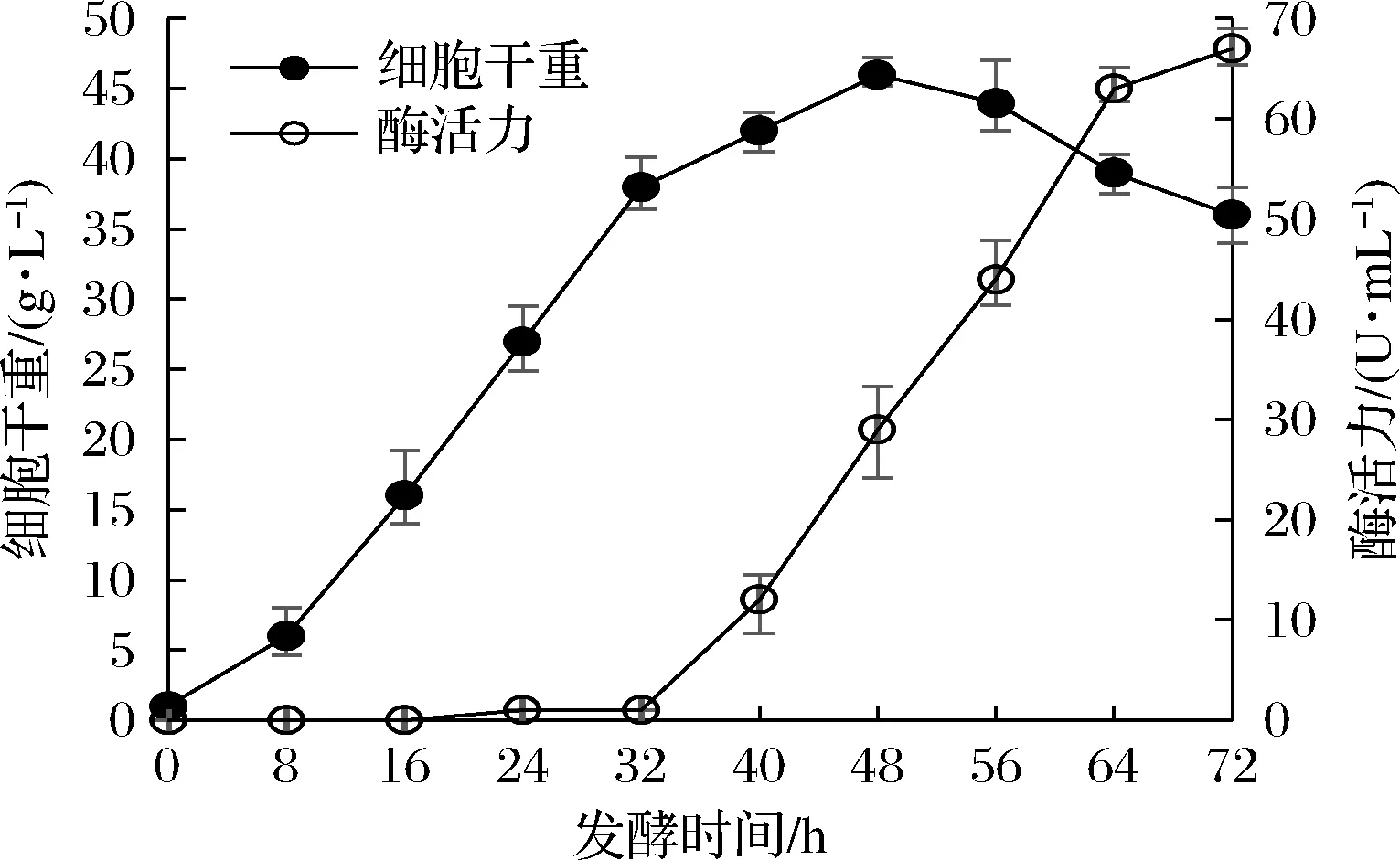
图6 分批补料发酵条件下的菌体生长与胞外酶活力增长曲线Fig.6 Cells growth and production of extracellular enzymatic activity
2.5 重组β-1,3-葡聚糖酶对灵芝细胞壁的裂解
将2.4中获得的BGL6906酶液与灵芝菌丝体细胞混合观察灵芝细胞的变化,发现即使使用高浓度的酶与少量灵芝细胞混合反应也无法观察到原生质体的形成。在前期工作中,Cellulosimicrobiumsp.6906发酵液10倍浓缩液与灵芝细胞混合反应30 min即可观察到明显的原生质体,而当时所使用的Cellulosimicrobiumsp.6906发酵液浓缩液的β-1,3葡聚糖酶酶活力只有4 U/mL。由此推测BGL6906可能需要和其他酶共同作用才能有效水解灵芝细胞壁。由于Cellulosimicrobiumsp.6906发酵过程需要使用寡聚茯苓多糖进行诱导,因此目前尚不具备工业化应用的可能性。为了寻找一种适合于工业应用的方法,将BGL6906酶液与市场上可以获得的各种工业酶制剂配合使用观察灵芝细胞壁裂解效果。结果发现,BGL6906与市售果胶酶或木瓜蛋白酶配合水解灵芝菌丝体细胞后可以观察到少量原生质体。结果如表2所示。

表2 BGL6906与其他酶制剂配合水解灵芝菌丝体的效果Table 2 Effects of cooperative hydrolysis of Ganoderma mycelium with BGL6906 and other enzyme products
由表2可见,BGL6906以及果胶酶和木瓜蛋白酶单独作用于灵芝菌丝体均不能有效水解细胞壁。BGL6906先水解再加果胶酶或木瓜蛋白酶可以水解灵芝细胞壁获得少量原生质体;但2种酶同时加入,或者是果胶酶或木瓜蛋白酶加入后再加BGL6906,均无法得到原生质体,出现这种现象最可能的原因是BGL6906被降解。木瓜蛋白酶本身就能够水解蛋白质,降解BGL6906导致其失去活性很容易理解。果胶酶则可能是生产过程中混入了蛋白酶。目前市售果胶酶一般采用黑曲霉发酵获得,黑曲霉也是酸性蛋白酶的生产菌,因此果胶酶发酵过程很可能伴随蛋白酶的生成,并最终进入果胶酶成品中。
为验证这一猜想,将BGL6906与果胶酶混合,大约5 min后混合液中β-1,3-葡聚糖酶的活性几乎检测不到,但果胶酶活性则基本不变,可见2种酶共同作用时BGL6906被降解的可能性很大。据此,将灵芝菌丝体用果胶酶水解后离心,去除上清液后再用BGL6906水解,可以获得少量的原生质体。在此结果的启发下,进一步采用以下方法水解灵芝细胞:用BGL6906水解灵芝细胞5 min后加入果胶酶水解30 min,离心去除上清液后悬浮细胞再次加入BGL6906水解30 min,处理后的灵芝原生质体形成率明显上升,结果如图7所示。经过BGL6906+果胶酶+BG6906处理的灵芝菌丝体虽然大部分没有变成原生质体,但菌丝形态发生了明显的变化,主要特点是菌丝变短、菌体膨大,这很可能是细胞壁强度弱化的结果。
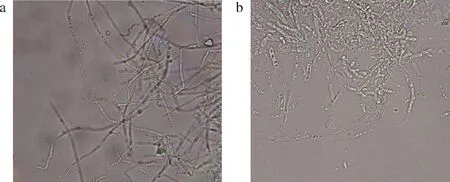
a-灵芝菌丝体;b-酶解后灵芝菌丝体图7 BGL6906和果胶酶处理前后细胞形态的差异Fig.7 Cell morphology before and after treatment with BGL6906 and pectinase
2.6 灵芝细胞裂解工艺的建立
从2.5的实验结果看,以BGL6906裂解灵芝细胞的关键是其与果胶酶交替作用。由于灵芝菌丝体粗壮,在实际生产中,一般采用板框过滤的方法进行细胞的收集。据此建立如下工艺:灵芝发酵到达终点时,用板框过滤收集菌丝体,收集完成后取出滤饼,用磷酸缓冲液重新悬浮菌体至与发酵液等体积,并加入BGL6906至终浓度为10 U/mL,反应5 min后加入果胶酶至终浓度为20 U/mL,反应10 min后再次压入板框过滤机过滤。为了尽量减少滤饼中果胶酶的残留,当菌液全部加入板框后,再向板框中压入相当于5倍发酵液体积的自来水对滤饼进行冲洗。滤饼取出后再次用磷酸缓冲液悬浮菌体,加入BGL6906至终浓度为20 U/mL,反应30 min后再加入果胶酶至终浓度为100 U/mL,反应30 min。通过上述工艺灵芝菌丝体经过2次BGL6909+果胶酶处理即经双酶2次处理。
经过上述处理的菌液镜检可以观察到少量原生质体形成。上述酶解的灵芝进一步用超声波进行细胞破碎,结果如图8所示。

图8 不同处理条件对超声波破碎效果的影响Fig.8 Effects of different treatment conditions on ultrasonic disruption注:未经酶水解实验中均使用灭活的酶;果胶酶水解实验中2次均使用灭活的BGL6906和有活性的果胶酶;单次双酶水解实验中,第一次双酶水解时使用灭活的酶,第二次双酶水解时均使用有活性的酶;两次双酶水解实验均采用有活性的酶。以上4组实 验均采用2次板框过滤和2次双酶水解
经过双酶2次处理的灵芝菌丝体经过5 min超声波处理后经过碱提法的蛋白提取率为76%,而对照组,即采用灭活的酶处理的细胞则经过30 min超声波处理后也只能得到约40%的蛋白。可见BGL6906和果胶酶交替处理可以极大的弱化细胞壁,提高超声波处理的效率。
上述工艺需要2次BGL6909-果胶酶处理菌丝体细胞,其中第一次处理时酶的用量和处理时间必须严格控制,否则在随后的板框过滤时菌液会堵塞滤布的滤孔而无法收集细胞。板框过滤的关键是滤液中的固形物互相堆积形成有孔隙的滤饼,灵芝菌丝体粗壮并有一定的刚性,这是其容易通过板框过滤进行细胞收集的关键原因。酶的过度处理可能导致一部分细胞的细胞壁弱化,从而致菌丝变得柔软而无法形成滤饼,同时堵塞滤饼和滤布的孔隙,因此第一次酶处理时酶解程度需要严格控制。
将经过碱提法得到的蛋白溶液进一步进行蛋白提取和初步纯化,获得富硒蛋白粉。检测结果显示,未经酶水解得到的蛋白中硒含量约为584.6 mg/kg,经2次双酶水解得到的蛋白中硒含量约为537.2 mg/kg。两者非常接近,可见2次双酶提取的方法对蛋白中硒的稳定性并没有明显的影响。本文采用的蛋白提取方法参考了邓乾坤等[18]建立的富硒酵母蛋白提取方法,并与该文献中得到的蛋白中硒含量(358.9 mg/kg)接近。
3 讨论
本研究对一种来源于纤维微菌的β-1,3-葡聚糖酶基因bgl6906进行了异源表达,获得的重组酶BGL6906与文献报道的纤维微菌属来源的β-1,3-葡聚糖酶基本一致[19-21]。BGL6906在性质上与已报道的上述β-1,3-葡聚糖酶的最大的差异是其只对茯苓多糖表现出高活性。以茯苓多糖为底物时,BGL6906的比酶活达到567.8 U/mg,以酵母聚糖、热凝胶和昆布多糖为底物时,比酶活力大约只有水解茯苓多糖的酶活力的1/7、1/24和1/35。纤维素纤维微菌和Cellulosimicrobiumfunkei来源的β-1,3-葡聚糖酶是目前有文献报道的与BGL6906氨基酸序列最接近的葡聚糖酶,三者的氨基酸同源性均在90%以上。纤维素纤维微菌和C.funkei来源的β-1,3-葡聚糖酶的最适底物分别是热凝胶和昆布多糖[20-21],可见纤维微菌属来源的β-1,3-葡聚糖酶即使氨基酸序列非常接近其最适底物也可能不完全相同。
β-1,3-葡聚糖酶主要在酵母细胞壁水解、生物防治以及寡聚葡聚糖制备等方面有一定的应用和研究。葡聚糖是灵芝细胞壁的主体成分之一,颜梦秋等[22]研究灵芝属真菌紫芝的细胞壁葡聚糖结构时发现,其葡聚糖主要由β-1,3-糖苷键连接而成。β-1,3-葡聚糖酶的重组酶在灵芝等大型子实体真菌的菌丝体裂解方面的应用研究未见报道。从本文研究结果看,BGL6906单独并不能有效水解灵芝细胞壁,这可能是限制这一类酶应用的原因。在对大型真菌的酶法破壁的研究中,黄敏等[23]发现,木瓜蛋白酶、纤维素酶、果胶酶等处理金针菇菌丝体细胞后有利于进一步的细胞破碎。本研究发现,木瓜蛋白酶和果胶酶与BGL6906有协同作用的效果。纤维素酶是有较多文献报道的能促进大型真菌菌丝体细胞壁降解的酶。本研究也发现,木霉来源的中性纤维素酶单独作用于灵芝菌丝体细胞时可以明显提高后续超声波破碎的效率,但与BGL6906联合作用时不管是共同使用还是交替使用均未见降解效果增强的现象。这可能是因为纤维素酶和BGL6906均作用于葡聚糖,功能上有相似性,因此不能产生协同作用。
4 结论
本文从1株纤维微菌属细菌中克隆了1条β-1,3-葡聚糖酶基因,表达产物重组酶BGL6906可以分泌到细胞外。重组酶BGL6906与果胶酶联合作用可以大幅度提高灵芝菌丝体的破壁效率。这一方法目前最大的缺陷是2种酶无法在同一体系中使用,原因可能是果胶酶中的蛋白酶降解BGL6906。今后工作中,采用同一个表达体系异源表达BGL6906和果胶酶可能有助于解决这一问题。
猜你喜欢
乡村科技(2021年12期)2021-09-06
化学与生物工程(2021年1期)2021-01-22
食品与机械(2020年8期)2020-09-18
中国糖料(2019年1期)2019-02-13
中国酿造(2018年10期)2018-11-03
中国野生植物资源(2018年3期)2018-08-06
中成药(2018年7期)2018-08-04
中国酿造(2016年12期)2016-03-01
安全(2015年7期)2016-01-19
中国果菜(2015年2期)2015-03-11
