
利用F2:3家系来源单倍体定位玉米雄穗相关性状QTL及全基因组选择
2023-01-16 05:06许加波吴鹏昊黄博文陈占辉马月虹任姣姣
作物学报 2023年3期
许加波 吴鹏昊 黄博文 陈占辉 马月虹 任姣姣
利用F2:3家系来源单倍体定位玉米雄穗相关性状QTL及全基因组选择
许加波 吴鹏昊 黄博文 陈占辉 马月虹 任姣姣*
新疆农业大学农学院, 新疆乌鲁木齐 830052
雄穗大小影响玉米光合作用合成的养分分配, 进而影响雌穗发育以及由此决定的穗行数、行粒数、结实率、百粒重等产量构成因素。本研究用优良自交系郑58和B73构建的F2:3家系诱导单倍体, 通过48K液相杂交探针捕获技术获得基因型, 结合多环境单倍体表型数据, 对雄穗相关性状采用完备区间作图法(inclusive composite interval mapping, ICIM)进行QTL (quantitative trait locus)定位, 采用(ridge regression best linear unbiased prediction, RRBLUP)模型探索全基因组选择中训练群体大小及SNP标记数目对预测精度的影响。结果表明, 雄穗主轴长、一级分枝数、二级分枝数和总分枝数遗传力分别为0.82、0.88、0.84和0.88。雄穗主轴长检测到2个QTL, 分别位于bin1.03和bin4.09, 表型贡献率为6.02%和11.10%。一级分枝数检测到2个QTL, 分别位于bin1.05和bin4.05, 表型贡献率为9.17%和11.75%。二级分枝检测到2个QTL, 分别位于bin2.03和bin3.06, 表型贡献率为5.51%和5.65%。总分枝数检测到2个QTL, 分别位于bin1.04和bin4.05, 表型贡献率为9.37%和10.83%。其中, 一级分枝数和总分枝数在bin4.05定位到了一个相同位点, 一因多效。全基因组选择五倍交叉验证的预测精度分别为0.36、0.41、0.28、0.38。当训练群体达到总群体60%时, 标记密度达到500个时, 即可以得到较高的预测精度。
玉米; 单倍体; 雄穗; QTL; 全基因组选择
玉米(L.)是典型的雌雄同株异花授粉作物, 玉米雄穗大小会影响到玉米籽粒的产量。如果雄穗过大, 不但会争抢玉米雌穗的养分, 而且会遮挡太阳光, 导致下部叶片光合作用受影响[1]; 如果雄穗过小, 又不能保证足够的花粉量供应, 不利于应对逆境条件下结实率降低的风险[2-4]。前人的研究指出玉米雄穗表型性状属于数量遗传性状, 并且容易受到外界因素的影响。但仅通过表型数据研究费时费力, 所以对控制玉米雄穗相关性状进行遗传研究有利于提高育种效率。
申涛等[5]利用T32和黄C两个自交系构建F2:3家系, 采用完备区间作图(ICIM)法对雄穗分枝数进行了QTL检测, 共检测到3个QTL, 分别位于4号、8号、9号染色体, 总共可解释39.41%的表型变异, 雄穗分枝数受加性(additive, A)、部分显性(partial dominant, PD)和超显性(over dominant, OD)控制。杨钊钊等[6]利用黄早4组配的11个重组自交系(recombinant inbred lines, RIL)群体, 发现雄穗主轴长和雄穗一级分枝数存在5个在3个自交RIL群体中稳定表达的区间, 位于2号、3号、6号和8号染色体上。高世斌等[7]在正常和干旱2个环境下, 对N87-1和9526组配的F3家系进行雄穗分枝数和雄穗主轴长QTL检测, 正常环境下在5号和7号染色体以及干旱环境下在2号、5号、7号和10号染色体定位到雄穗分枝数相关位点, 在正常环境下于2号和6号染色体以及干旱环境下于2号、4号和10号染色体检测到雄穗主轴长相关QTL相关位点。其中雄穗分枝数遗传作用方式以部分加性为主, 雄穗主轴长全部表现为显性和超显性。张先创[8]在1号、4号、5号和8号染色体上定位到雄穗分枝数QTL位点, 在3号、5号、7号、9号和10号染色体定位到雄穗主轴长相关位点, 但所有位点的表型贡献率均小于6.26%。Wu等[9]使用ZHL908和HCL645构建的F2:3家系, 在2号、3号、6号、7号和10号染色体定位到雄穗主轴长相关QTL位点, 在1号、2号、9号和10号染色体定位到雄穗一级分枝数相关QTL位点。Upadyayula等[10]对IHO (illinois high oil)和B73两个自交系构建的BC1S1系进行研究, 在5号和7号染色体定位到控制一级分枝数的QTL位点, 在1号、4号、5号、6号和7号染色体定位到雄穗主轴长的QTL位点。Mickelson等[11]使用B73和Mo17两个玉米自交系组建的重组自交系对雄穗分枝数进行QTL定位, 定位结果分布于1号、2号、3号和4号染色体。Liu等[12]使用Lv28与H082183两个玉米自交系构建的F2:3家系, 定位到7个雄穗分枝数相关QTL位点, 分别位于1号、2号、3号、4号和8号染色体。尽管目前关于玉米雄穗相关性状的遗传研究很多, 但共定位的位点较少, 仍需要对不同群体和不同环境下的定位结果进行探索, 挖掘在多群体、多环境下稳定的QTL。
全基因组选择(genomic selection, GS)可以提高优良基因型的选择效率, 缩短育种周期。与分子标记辅助选择相比, GS在提高复杂性状的遗传增益方面有明显的优势[13]。植物全基因组选择育种技术通过训练群体收集表型数据和基因型数据, 使用特定的模型估计分子标记效应值或个体育种值, 再根据预测群体的基因型数据和模型拟合结果对待测群体的表型值进行预测[14]。全基因组选择预测精度受多种因素影响, 如预测模型种类、训练群体大小、训练群体与预测群体亲缘关系、标记密度、连锁不平衡等。目前已经有玉米方面的应用, Riedelsheimer等[15]对285个不同马齿型自交系玉米的产量、株高和雌花吐丝期等7个性状进行研究, 利用SNP标记和岭回归最佳线性无偏预测(RRBLUP)模型, 采用五倍交叉验证法进行全基因组选择研究, 结果显示7个性状的预测准确度均较高, 分布在0.72到0.81之间。Liu等[16]选用1个自然群体和4个双亲群体, 对株高、穗位高、穗长、穗粗、单株产量和百粒重进行全基因组选择预测精度比较, 发现预测精度在训练群体为预测群体3倍时可达到较高预测精度。
前人关于玉米雄穗性状的遗传研究都是基于二倍体群体, 目前无基于单倍体群体进行雄穗基本性状的研究。单倍体和二倍体在遗传上存在倍性差异, 对单倍体家系进行研究在遗传定位角度可能会得出互补的结论。本研究利用来源于B73与郑58两个玉米自交系构建的F2:3家系来源的单倍体, 在多环境下采集玉米雄穗主轴长、雄穗一级分枝数、雄穗二级分枝数和雄穗总分枝数表型数据, 结合基因型数据对玉米单倍体雄穗相关性状进行QTL定位, 解析雄穗相关性状遗传机制, 并进行全基因组选择分析, 研究不同训练群体大小和不同SNP标记个数对预测精度的影响, 为GS在育种中应用提供理论依据。
1 材料与方法
1.1 试验材料及田间设计
选取B73和郑58两个玉米自交系作为亲本, 通过组配得到200个F2:3家系。利用农大高油高诱诱导系CHOI3花粉对2个亲本和F2:3家系进行授粉诱导单倍体, 通过R1-nj颜色标记挑选单倍体。2021年将单倍体材料分别于4月30日种植于新疆昌吉市九圣禾产业园, 5月10日和5月17日种植于新疆呼图壁实验站(原中国农业大学教授工作站), 共3个环境。每个环境设置2个重复, 根据每个家系单倍体的种子量种植3~5行, 行长2.5 m, 行距50 cm, 株距10 cm。
由于R1-nj颜色标记鉴别的准确率很难达到100%, 需要根据单倍体和二倍体的田间表型对单倍体进行2次鉴定。玉米单倍体植株矮小, 叶片窄, 叶片尖端向主茎聚拢, 主茎较细, 长势较弱。玉米二倍体植株高大, 叶片宽, 叶尖端片弯曲下垂, 主茎较粗, 长势较强。一般于六至八叶期进行二倍体除杂工作。
1.2 表型鉴定
单倍体散粉结束后, 每个家系内随机选择10株进行表型调查, 测定雄穗主轴长(tassel length, TL)、雄穗一级分枝数(tassel primary branch number, TPBN)、雄穗二级分枝数(tassel secondary branch number, TSBN)和雄穗总分枝数(tassel branch number, TBN)。鉴定方法如表1。

表1 玉米单倍体雄穗相关性状测定方法
1.3 表型数据分析
利用META-R软件(http://hdl.handle.net/11529/ 10201), 使用混合线性模型对玉米雄穗相关性状表型数据进行分析, 获得最佳线性无偏估计(best linear unbiased prediction, BLUP)值, 用于后续的表型分析、相关性分析、QTL定位及全基因组选择分析。采用Pearson correlation对单倍体雄穗相关性状进行相关性分析。利用R软件对原始数据进行双因素方差分析和遗传力分析。广义遗传力计算公式:
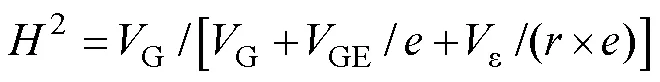
式中,2是广义遗传力,G是遗传方差,GE为基因与环境互作方差,V为残差方差,为重复数,为环境个数。
1.4 基因型分析
将F2群体和亲本种植后1个月时取叶片, 采取十六烷基三甲基溴化铵(hexadecyl trimethyl ammonium bromide, CTAB)法提取DNA, 由中玉金标记(北京)生物技术股份有限公司采用48K液相杂交探针捕获技术进行基因型检测。使用BWA软件将检测到的原始基因型数据与B73_RefGen_v4_genomic参考基因组进行比对。使用vcftools进行过滤, 亲本过滤参数为: GQ大于30, Q大于30, 缺失率小于0.97; 子代过滤参数为: GQ大于10, Q大于30, 缺失率小于0.97。将亲本和子代数据合并后共得到62,504个位点用于GS分析。再对标记进行多态性过滤、标记偏分离过滤(>0.05), 采用滑动窗口法构建Bin标记[19], 共得到1775个Bin标记用于遗传图谱构建。
1.5 遗传图谱构建和QTL定位
选择QTL IciMapping v4.2软件中MAP功能构建遗传连锁图谱, 采用BIP功能选择完备区间作图法对4个雄穗相关性状进行QTL定位, LOD值设置为2.5。定位结果可视化选用Mapchart 2.32软件。按Stuber等[20]的标准度量确定QTL位点的遗传作用方式, 将定位结果中显性效应值与加性效应值相除后求得绝对值获得显性度, 按显性度大小分类为: 加性(A): 显性度=0~0.20; 部分显性(PD): 显性度= 0.21~0.80; 显性(dominant, D): 显性度=0.81~1.20; 超显性(OD): 显性度>1.20。
1.6 全基因组选择研究
对单倍体雄穗性状全基因组选择分析, 选用62,504个SNP标记, 多环境BLUP值作为表型值, 选用RRBLUP模型对单倍体雄穗相关性状进行全基因组选择分析。采用5倍交叉验证法, 即将群体随机分成5等份, 选取其中4份为训练群体(同时具有基因型值和表型值), 1份为预测群体(只有基因型值), 进行全基因组选择。使用R软件的rrBLUP包对预测群体育种值进行预测[21], 获得全基因组估计育种值, 重复100次。GS预测精度为预测群体真实育种值与全基因组估计育种值的相关系数[22]。
为了探究不同训练群体大小对全基因组选择预测精度的影响, 采用全部62,504个SNP标记对雄穗相关性状进行GS, 训练群体大小设置从10%到90%以10%的梯度由小到大递增, 剩下群体为预测群体进行GS, 每个训练群体大小重复100次。为了探究SNP标记数目对GS预测精度的影响, 分别选择10、30、50、100、300、500、1000、3000、5000、10,000和50,000的标记数目, 采用五倍交叉验证进行GS, 每个标记密度重复100次[23]。
2 结果与分析
2.1 单倍体雄穗相关性状的表型分析
郑58单倍体雄穗主轴长平均为17.90cm, 一级分枝数平均为5.93, 二级分枝数平均为0.85, 总分枝数平均为6.78。B73单倍体雄穗主轴长平均为19.65cm; 一级分枝数平均为7.57; 二级分枝数平均为1.12; 总分枝数平均为5.92。郑58单倍体雄穗主轴长及雄穗一级分枝数显著低于B73单倍体。
由表2可知, 单倍型雄穗的主轴长度平均值为18.52cm, 分布范围为17.33~19.90cm; 雄穗一次分枝数平均为5.85, 分布范围为3.79~8.32; 雄穗二级分枝数平均为0.79, 分布范围为0.50~1.23; 雄穗总分枝数平均为6.64, 分布范围为4.33~9.51。从变异系数结果来看, 除雄穗主轴长之外其余变异系数均较大, 存在广泛的遗传变异。所有性状的偏度和峰度的绝对值均小于1, 服从正态分布, 符合数量性状遗传特征。
2.2 单倍体雄穗相关性状方差分析
表3表明, 玉米单倍体雄穗4个性状在基因型间均为极显著差异(<0.01)。在环境间除雄穗主轴长表现为显著差异(<0.05)之外, 其余均表现为极显著差异(<0.01)。在基因型与环境互作间, 雄穗一级分枝数表现为极显著差异(<0.01), 雄穗总分枝数为显著差异(<0.05), 其余2个性状差异均不显著。雄穗主轴长、雄穗一级分枝数、雄穗二级分枝数和雄穗总分枝数的遗传力分别为0.82、0.88、0.84和0.88, 遗传力均较高, 说明4个单倍体雄穗相关性状主要受遗传因素控制。
2.3 单倍体雄穗相关性状相关性分析
图1相关性分析结果表明, 雄穗主轴长与雄穗分枝数3个性状的相关系数均小于0.25。雄穗分枝数相关的3个性状存在极显著正相关, 雄穗一级分枝数与雄穗二级分枝数的相关性系数为0.58 (<0.001), 穗二级分枝数与雄穗总分枝数的相关系数为0.70 (<0.001), 雄穗一级分枝数与雄穗总分枝数的相关系数为0.99 (<0.001)。
2.4 遗传连锁图谱的构建
图2所示, 1775个bin标记构建的遗传连锁图谱总长度为866.05cM, 标记间的平均遗传距离为0.49cM。所有染色体上的标记数均超过100个, 其中1号染色体上的标记数最多为316个, 6号染色体上的标记数最少为105个。
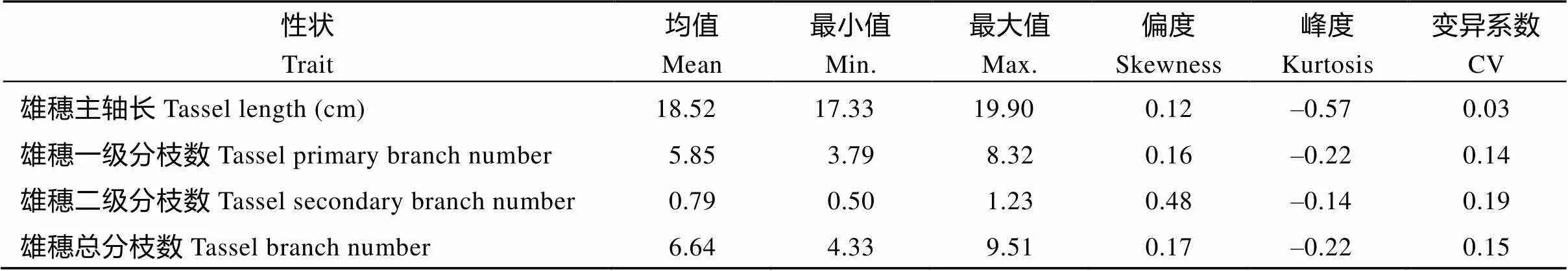
表2 玉米单倍体雄穗相关性状描述性统计
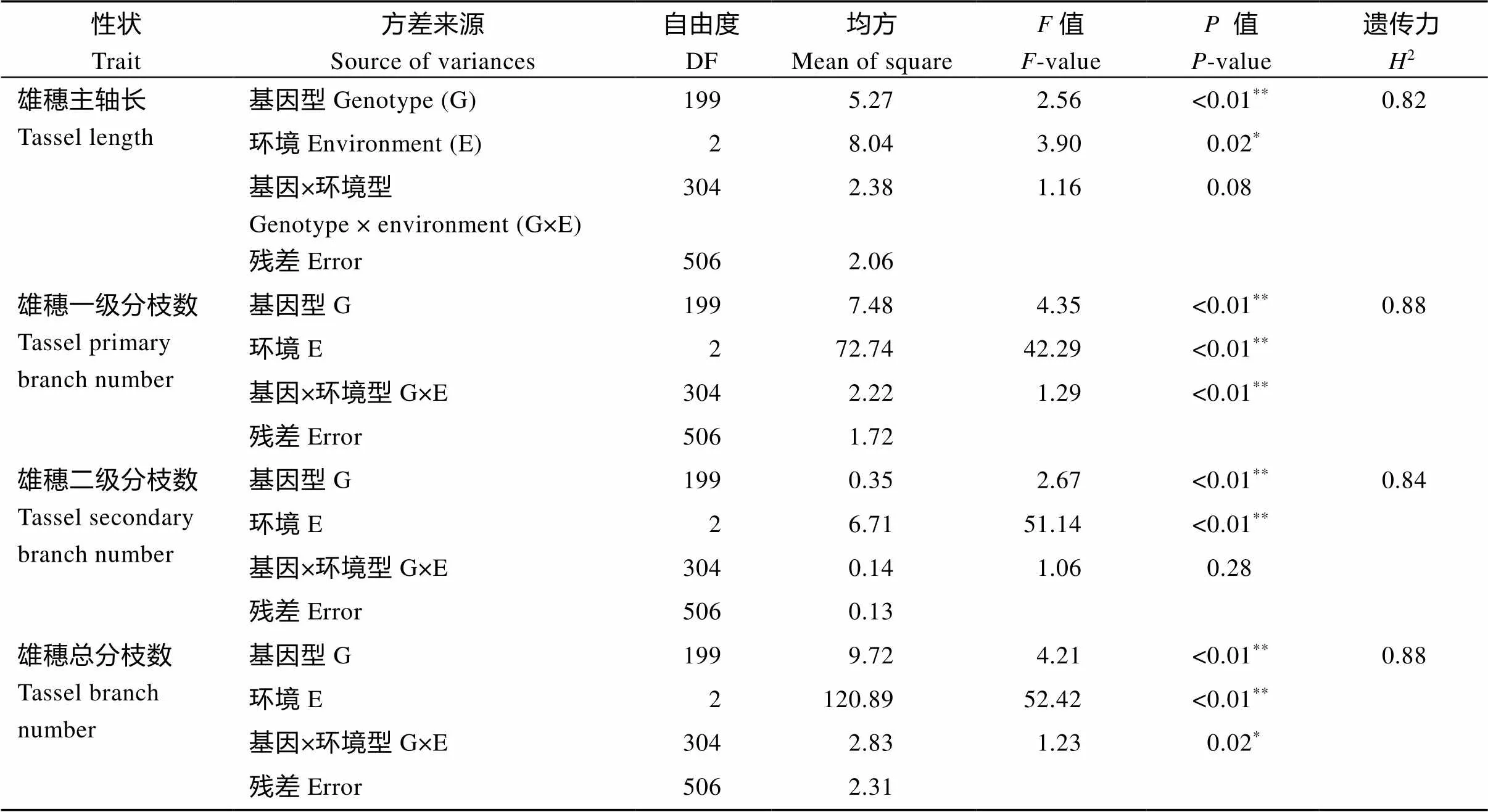
表3 玉米单倍体雄穗相关性状的方差及广义遗传力分析
*在0.05水平上差异显著;**在0.01水平上差异显著。*:< 0.05;**:< 0.01.

图1 玉米单倍体雄穗相关性状的相关性分析
*在0.05水平上差异显著; **在0.01水平上差异显著; ***在0.001水平上差异显著。圆形由小到大代表相关系数由低到高。
*:< 0.05;**:< 0.01; ***:< 0.001. The circle from small to large represents the correlation coefficient from low to high.
2.5 QTL定位
由图2和表4所示, 雄穗主轴长定位到2个QTL位点, 分别位于bin1.03和bin4.09。1号染色体的位于32.50~32.53 Mb (B73_RefGen_v4_ genomic), LOD值为3.01, 表型贡献率为6.02%, 加性效应为0.05, 增效等位基因由母本郑58提供, 遗传作用方式为超显性。4号染色体的位于223.30~224.37 Mb, LOD值为5.40, 表型贡献率为11.10%, 加性效应为0.27, 增效等位基因由母本郑58提供, 遗传作用方式为加性。
雄穗一级分枝定位到2个QTL位点, 分别位于bin1.05和bin4.05。1号染色体的位于102.35~105.19 Mb, LOD值为4.98, 表型贡献率为9.17%, 加性效应为0.37, 增效等位基因由母本郑58提供, 遗传作用方式为部分显性。4号染色体的位于75.57~79.81 Mb, LOD值为6.29, 表型贡献率为11.75%, 加性效应为–0.42, 增效等位基因由父本B73提供, 遗传作用方式为部分显性。
雄穗二级分枝定位到2个QTL位点, 分别位于bin2.03和bin3.06。其中2号染色体的位于26.29~27.50 Mb, LOD值为2.69, 表型贡献率为5.51%, 加性效应为–0.06, 增效等位基因由父本B73提供, 遗传作用方式为加性。3号染色体的位于185.21~185.33 Mb, LOD值为2.78, 表型贡献率为5.65%, 加性效应为0.06, 增效等位基因由母本郑58提供, 遗传作用方式为加性。
雄穗总分枝数定位到2个QTL位点, 分别位于bin1.04和bin4.05。1号染色体的位于56.92~57.40 Mb, LOD值为5.11, 表型贡献率为9.37%, 加性效应为0.42, 增效等位基因由母本郑58提供, 遗传作用方式为部分显性。4号染色体的位于75.57~79.81 Mb, LOD值为5.80, 表型贡献率为10.83%, 加性效应为–0.46, 增效等位基因由父本B73提供, 遗传作用方式为部分显性。此位点与定位区间一致, 为一因多效位点。
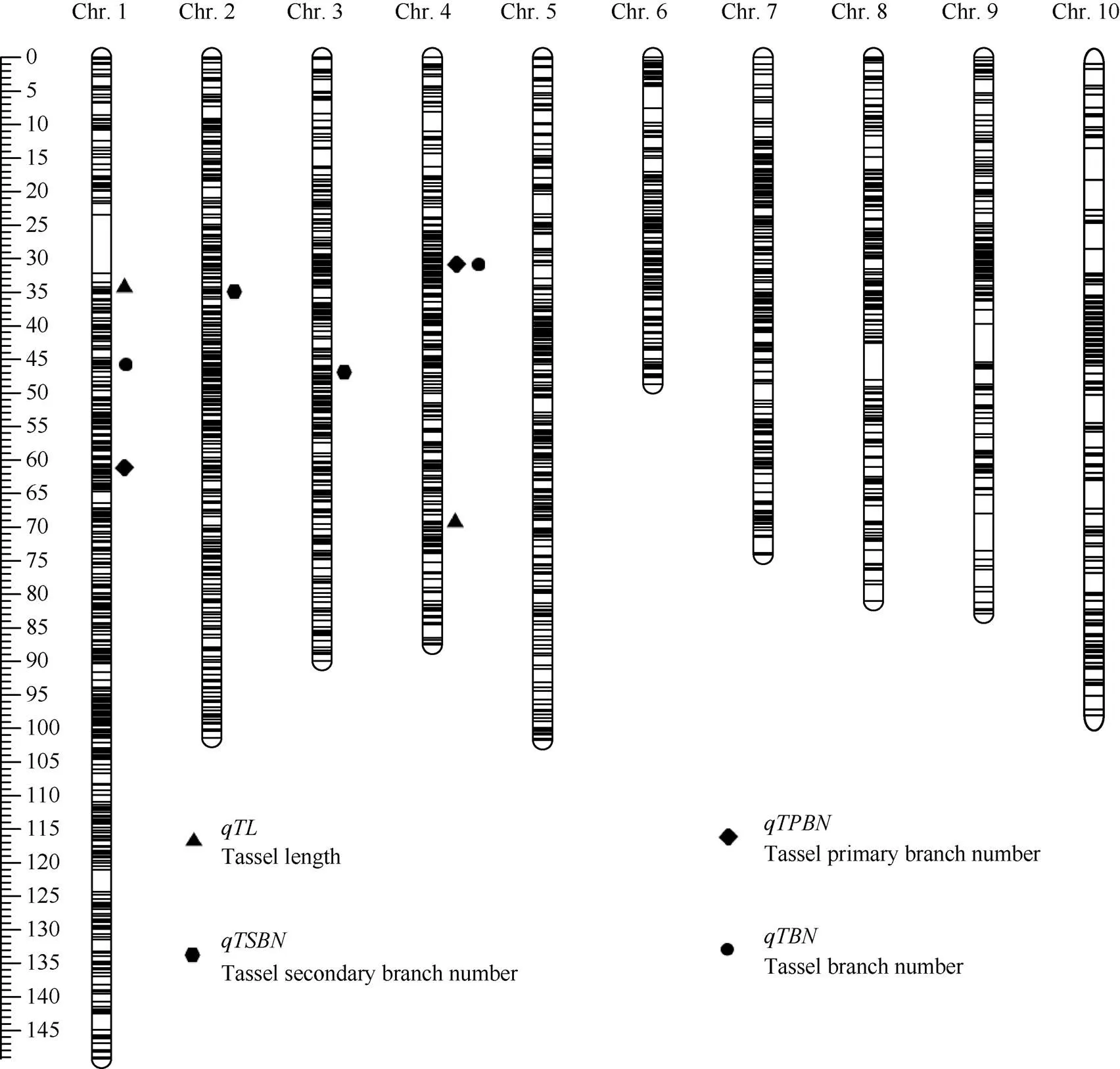
图2 玉米单倍体群体雄穗相关性状的QTL定位
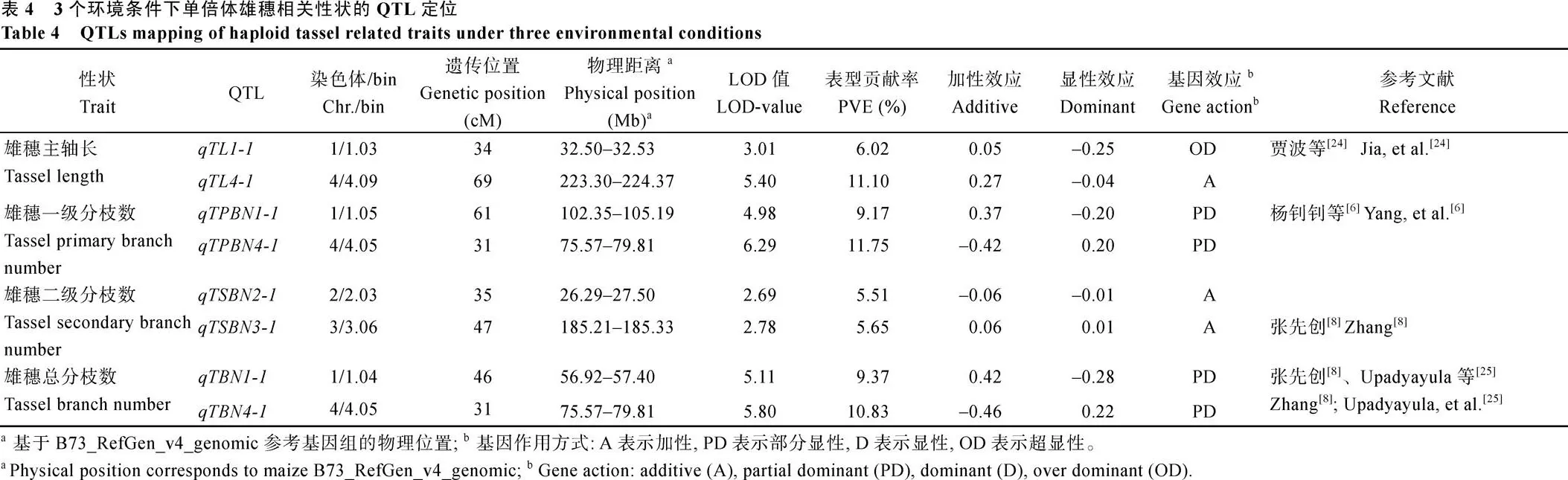
2.6 全基因组选择
GS结果如图3所示, 雄穗主轴长、雄穗一级分枝数、雄穗二级分枝数和雄穗总分枝数预测精度分别为0.36、0.41、0.28和0.38。其中雄穗一级枝数GS预测精度变异幅度最大, 雄穗二级分枝数GS预测精度最低。
由图4可知, 随着训练群体逐渐增大, 4个性状GS预测精度均呈现先上升后稳定的趋势。当训练群体由10%上升到60%时, 雄穗主轴长、雄穗一级分枝数、雄穗二级分枝数和雄穗总分枝数4个性状都达到相对较高的预测精度, 分别为0.33、0.39、0.25和0.38。当训练群体超过60%后, 4个性状预测精度增加幅度很小。
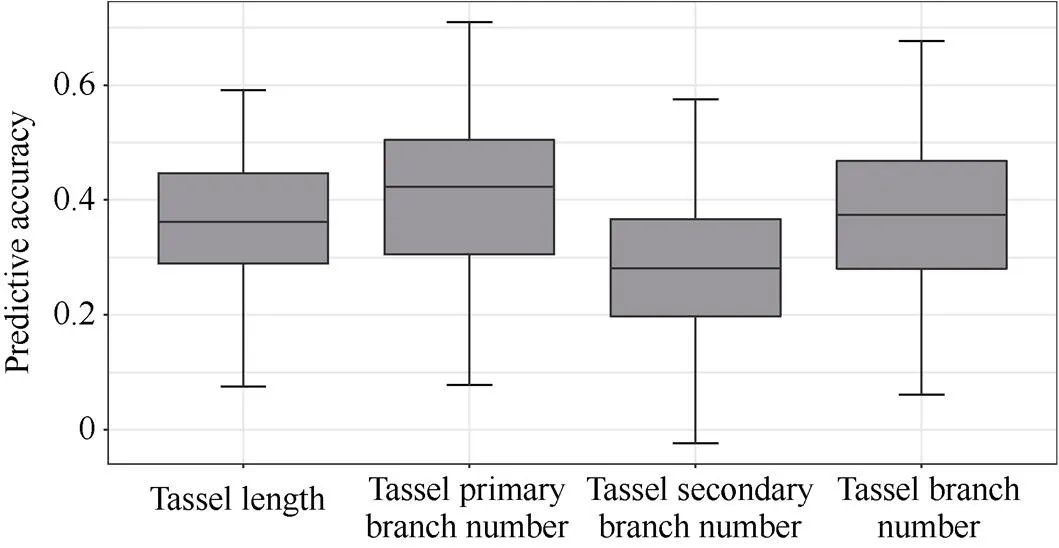
图3 玉米单倍体群体雄穗相关性状全基因组选择预测精度
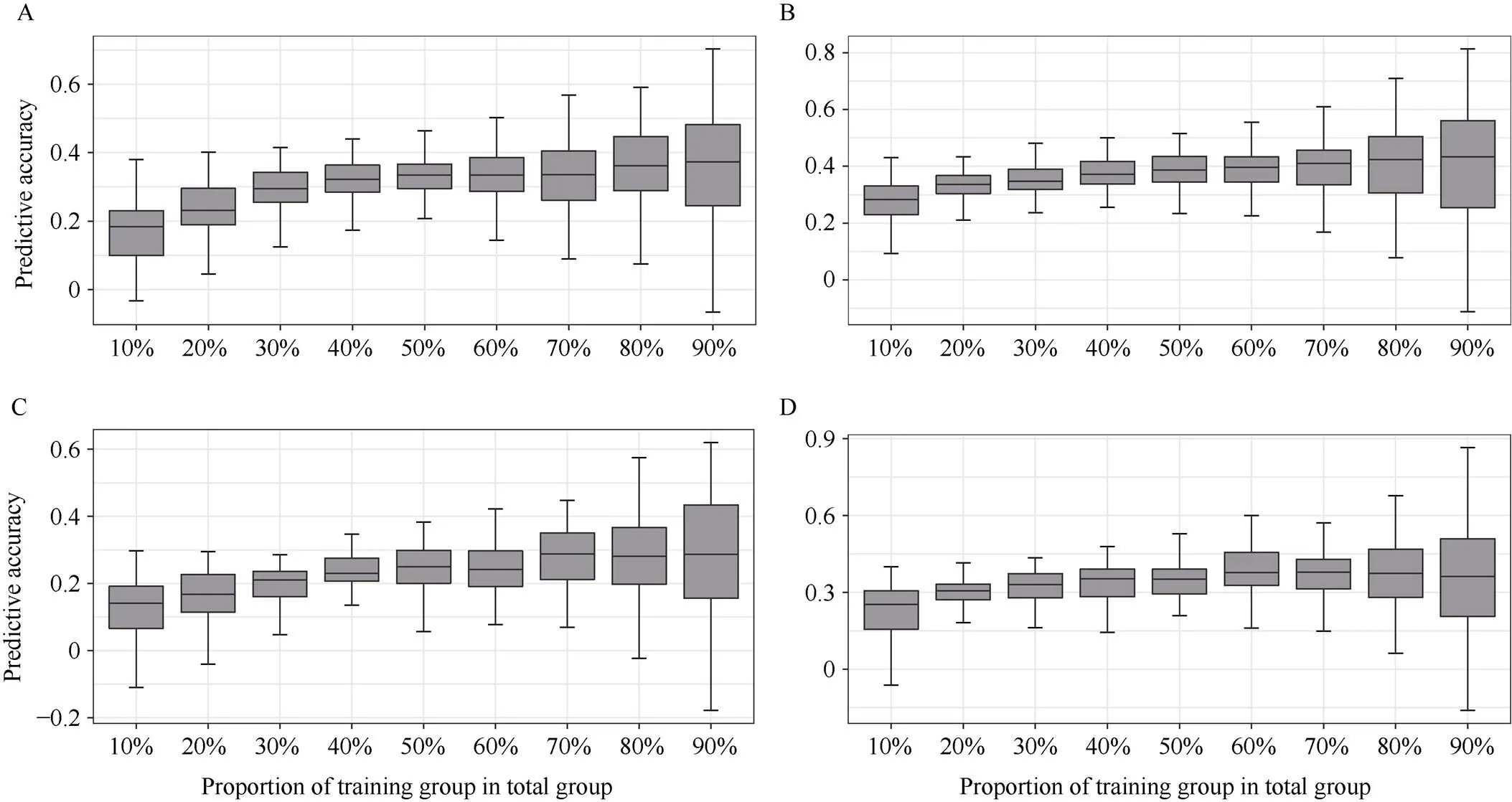
图4 玉米单倍体不同训练群体大小雄穗相关性状全基因组选择预测精度
A: 雄穗主轴长; B: 雄穗一级分枝数; C: 雄穗二级分枝数; D: 雄穗总分枝数。
A: tassel length; B: tassel primary branch number; C: tassel secondary branch number; D: tassel branch number.
由图5可知, 随着SNP标记数逐渐增多, 4个性状GS预测精度在标记数目10到500内呈现上升趋势, 变异幅度逐渐减小。当SNP标记数目达到500个时, 雄穗主轴长、雄穗一级分枝数、雄穗二级分枝数和雄穗总分枝数4个性状均可以获得较高的预测精度, 预测精度分别为0.36、0.41、0.28和0.38。当SNP标记数超过500个后, 4个性状预测精度无明显提高。
3 讨论
玉米为雌雄同株异花, 雄穗大小会影响到玉米的产量。为了达到既能保证足够量的花粉供应, 又能兼顾提高光合作用、减少能量消耗的目的, 需要通过育种技术对其改良。前人采用F2群体对数量性状进行遗传研究时, 多采用F2:3家系的表型作为F2群体表型值进行分析。本研究对F2:3家系来源的单倍体进行表型的测量, 单倍体不存在同源染色体, 一个位点后代只有2种不同的基因型, 能够更好地代表对应F2单株的表型。本研究对郑58和B73构建的F2:3家系单倍体雄穗相关性状, 包括雄穗主轴长、雄穗一级分枝数、雄穗二级分枝数和雄穗总分枝数进行方差分析, 4个雄穗性状的遗传力为0.82~0.88, 遗传力较高, 与前人研究结果一致[6]。杨钊钊等[6]对以黄早四为共同亲本组配的11个RIL群体雄穗一级分枝数、雄穗主轴长进行遗传力分析, 雄穗一级分枝数遗传力为0.92~0.96; 雄穗主轴长遗传力为0.85~0.90。该结果说明这些性状受遗传因素影响较大, 对玉米雄穗相关性状进行选择育种时, 可以在早代进行选择, 达到定向选择的目的。而且对其进行遗传研究有利于分子标记辅助选择, 提高育种效率。

图5 玉米单倍体群体不同SNP个数雄穗相关性状全基因组选择预测精度
A: 雄穗主轴长; B: 雄穗一级分枝数; C: 雄穗二级分枝数; D: 雄穗总分枝数。
A: tassel length; B: tassel primary branch number; C: tassel secondary branch number; D: tassel branch number.
雄穗一级分枝数在1号染色体的bin1.05区间定位到及4号染色体的bin4.05区间定位到, 共解释20.92%的表型变异。雄穗总分枝数在1号染色体bin1.04和4号染色体bin4.05区间分别定位到与, 共解释20.20%的表型变异。其中与的定位区间一样, 是同一个QTL位点, 这可能是因为雄穗一级分枝数和雄穗总分枝数极显著相关, 相关系数高达0.99。今后可以对一因多效位点加以利用来减少表型数据测定的工作量, 仅需要挑选其中一个性状即可以得到一致的结果。
前人关于雄穗一级分枝数和雄穗总分枝数的遗传研究较多, 本研究定位到的(bin1.04)、(bin4.05)和(bin4.05)位点在雄穗分枝数相关研究中均有报道[6,8,25]。杨钊钊等[6]利用黄早四和威风322组配RIL群体, 在bin4.05~4.06区间检测到雄穗一级分枝数主效QTL。Upadyayula等[25]利用Illinois Low Protein×B73构建的S1家系检测到2个控制雄穗总分枝数QTL, 其中一个位于bin4.05。张先创[8]对EHel与B73构建的F2:3家系进行研究, 检测到的位于bin1.04~1.05和bin4.05区间的和与本研究检测到的和在同一个区间。总之,与在不同遗传背景、不同倍性、不同环境、不同标记下均可检测到, 是控制雄穗总分枝数和雄穗一级分枝数的稳定QTL位点, 对其进行精细定位及候选基因的挖掘, 对揭示雄穗分枝数遗传机制具有重要意义。位点是本研究新检测到的位点, 为雄穗分枝数的遗传研究提供了新的QTL位点。前人对雄穗分枝数的研究多集中在雄穗一级分枝数和雄穗总分枝数的研究, 关于雄穗二级分枝数的遗传研究鲜有报道。本研究共检测到2个控制雄穗二级分枝数的QTL位点, 表型贡献率分别为5.51%和5.65%, 表型贡献率低, 说明雄穗二级分枝数是由微效多基因控制, 在育种时对此性状进行全基因组选择效果要优于分子标记辅助选择。
本研究共检测到2个控制雄穗主轴长的QTL位点,位于1号染色体bin1.03与位于4号染色体的bin4.09。贾波等[24]利用Z58和Y915构建的192个F2:3家系在淮安和扬州对雄穗长进行QTL定位, 共检测到15个QTL位点, 其中位于bin4.09的QTL与本研究其在同一区间, 贡献率为9.05%。
本试验虽然是利用单倍体进行遗传研究, 但与前人使用二倍体进行遗传研究的结果有部分重合位点(表4), 说明此类位点可以在不同群体中稳定表达, 可用于精细定位和基因克隆。同时定位结果与前人多数已挖掘位点存在不同, 与Wang等[26]、Yang等[27]、Xu等[28]和Chen等[29]研究结果不存在重合。原因可能是由于单倍体与二倍体定位结果存在定位结果的互补, 同时也由于试验材料的来源不同导致与前人已挖掘出的多数位点存在差异。由于数量性状同时受主效基因控制和受微效基因控制[30],建议在正向育种中使用基于QTL的分子标记辅助选择, 以提高早期选择周期中具有强加性QTL性状的等位基因频率, 而基于GS的分子标记辅助选择可用于更成熟的育种项目, 以额外捕获具有较小加性效应的等位基因[31]。
Lorenzana等[32]研究表明全基因组选择可以有效提高遗传增益。随着当今计算能力的不断提升使得数据库和智能算法更加全面, 并且新一代的测序技术成本越来越低, 从而提高了全基因组选择预测准确性[33]。本试验研究不同训练群体大小对GS预测精度影响时发现, 当训练群体占总群体的50%~60%时预测精度较好, 与Cao等[23]和Guo等[34]的研究结果一致。Cao等[23]利用1个自然群体和3个双亲DH群体对玉米鱼眼斑病(tar spot complex)抗性进行GS研究, 结果显示训练群体的最佳规模为群体总数的50%~60%。Guo等[34]利用不同训练群体大小对玉米籽粒锌含量全基因组选择预测精度进行研究, 结果表明自然群体DTMA (drought tolerant maize for Africa)、DH1和DH2三个群体在训练群体占总体50%~60%时, 均可达到比较高的预测精度。以上研究表明, 在进行全基因组选择育种时, 无论是自然群体还是双亲后代群体, 训练群体达到总群体的50%~60%时可得到较高的预测精度。
在研究不同标记数目对GS预测精度的影响时, 发现预测精度随着标记数目的增加呈现先上升后平稳的趋势, 当标记数目达到500个时预测精度较好, 该趋势与Cao等[35]、Ren等[36]和Liu等[37]]的研究结果一致, 但达到较好预测精度所需的最少分子标记数存在差异。Cao等[35]对自然群体和DH群体进行GS研究, 结果显示SNPs最佳数目分别为3000和500时可以获得良好预测精度。Ren等[36]]对玉米普通锈病抗性进行全基因组选择研究, 发现在自然群体中标记密度达到5000和双亲后代DH群体中标记密度达到300时, 预测精度达到了平台。Liu等[37]在在双亲群体中选择标记数目为20~500个时, rMG表现出显著增强。上述研究表明, 对自然群体进行GS时所需的标记数目为3000~5000个, 对双亲后代群体进行GS时需要的标记数为300~500个。群体最佳标记数不同可能是由连锁不平衡引起的, 自然群体连锁不平衡值小, 达到高预测精度所用到的分子标记数目多, 双亲后代群体连锁不平衡值大, 达到高预测精度所用到的分子标记数目少。本研究结果可为玉米雄穗相关性状的全基因组选择育种提供指导。
4 结论
本研究对玉米单倍体4个雄穗相关性状进行QTL定位, 这4个性状均在基因型、环境变异型具有显著差异, 且雄穗一级分枝数和雄穗总分枝数在基因型和环境互作变异项具有显著差异。广义遗传力分别为0.82、0.88、0.84和0.88。每个性状共定位到2个QTL位点, 其中位于bin4.05的和是同一个位点, 同时控制雄穗总分枝数和雄穗一级分枝数。全基因组选择结果表明, 在本研究的双亲后代群体中, 当训练群体占总体60%、SNP标记达到500个时, 全基因组预测精度较高, 进一步增大训练群体或增加标记数量, 预测精度无明显提高。本研究为解析玉米雄穗主轴长和雄穗分枝的遗传基础和GS育种提供了重要理论依据。
[1] Lambert R J, Johnson R R. Leaf angle, tassel morphology, and the performance of maize hybrids 1., 1978, 18: 499–502.
[2] 王赛, 王宇宇, 王石磊, 徐梦真, 邹欢, 侯清桂, 毛棣, 田磊, 陈彦惠, 吴连成. 基于SNP遗传图谱定位玉米雄穗分枝数和主轴长QTLs. 河南农业大学学报, 2019, 53: 671–676.
Wang S, Wang Y Y, Wang S L, Xu M Z, Zou H, Hou Q G, Mao D, Tian L, Chen Y H, Wu L C. QTLs mapping of tassel branch number and tassel total length in maize based on SNP genetic map., 2019, 53: 671–676 (in Chinese with English abstract).
[3] Wartha C A, Cargnelutti Filho A, Lúcio A D, Follmann D N, Kleinpaul J A, Simões F M. Sample sizes to estimate mean values for tassel traits in maize genotypes., 2016, 15: gmr15049151.
[4] Qin X, Tian S, Zhang W, Dong X, Ma C, Wang Y, Yan J, Yue B. Q Dtbn1, an F-box gene affecting maize tassel branch number by a dominant model., 2021, 19: 1183–1194.
[5] 申涛, 谭康, 李春红, 杨梅, 胡小兰, 蒋滔, 张志, 邱红波. 玉米株型相关性状的QTL定位. 分子植物育种, 2022, 20: 155–162.
Shen T, Tan K, Li C H, Yang M, Hu X L, Jiang T, Zhang Z, Qiu H B. QTL mapping for plant type related traits in maize., 2022, 20: 155–162 (in Chinese with English abstract).
[6] 杨钊钊, 李永祥, 刘成, 刘志斋,李春辉, 李清超, 彭勃, 张岩, 王迪, 谭巍巍, 孙宝成, 石云素, 宋燕春, 王天宇, 黎裕. 基于多个相关群体的玉米雄穗相关性状QTL分析. 作物学报, 2012, 38: 1435–1442.
Yang Z Z, Li Y X, Liu C, Liu Z Z, Li C H, Li Q C, Peng B, Zhang Y, Wang D, Tan W W, Sun B C, Shi Y S, Song Y C, Wang T Y, Li Y. QTL analysis of tassel-related traits in maize (L.) using multiple connected populations., 2012, 38: 1435–1442 (in Chinese with English abstract).
[7] 高世斌, 赵茂俊, 兰海, 张志明. 玉米雄穗分枝数与主轴长的QTL鉴定. 遗传, 2007, 29: 1013–1017.
Gao S B, Zhao M J, Lan H, Zhang C M. Identification of QTL associated with tassel branch number and total tassel length in maize., 2007, 29: 1013–1017 (in Chinese with English abstract).
[8] 张先创. 玉米雄穗相关性状的QTL定位. 西南大学硕士学位论文, 重庆, 2020.
Zhang X C. QTL Mapping of Tassel Related Traits in Maize (L.). MS Thesis of Southwest University, Chongqing, China, 2020 (in Chinese with English abstract).
[9] Wu X, Guo X Y, Wang A G, Liu P F, Wu W Q, Zhao Q, Zhao M Y, Zhu Y F, Chen Z H. Quantitative trait loci mapping of plant architecture-related traits using the high-throughput genotyping by sequencing method., 2019, 215: 212.
[10] Upadyayula N, Wassom J, Bohn M O, Rocheford T R. Quantitative trait loci analysis of phenotypic traits and principal components of maize tassel inflorescence architecture., 2006, 113: 1395–1407.
[11] Mickelson S M, Stuber C S, Senior L, Kaeppleret S M. Quantitative trait loci controlling leaf and tassel traits in a B73×Mo17 population of maize., 2002, 42: 1902–1909.
[12] Liu X, Hao L, Kou S, Su E, Zhou Y, Wang R, Mohamed A, Gao C, Zhang D, Li Y, Li H, Song Y, Shi Y, Wang T, Li Y. High-density quantitative trait locus mapping revealed genetic architecture of leaf angle and tassel size in maize., 2019, 39: 7.
[13] Olatoye M O, Clark L V, Wang J P, Yang X P, Yamada T, Sacks E L, Lipka A E. Evaluation of genomic selection and marker- assisted selection in Miscanthus and energy cane., 2019, 39: 1–16.
[14] 刘策, 孟焕文, 程智慧. 植物全基因组选择育种技术原理与研究进展. 分子植物育种, 2020, 18: 5335–5342.
Liu C, Meng H W, Cheng Z H. Plant genome-wide selection breeding technical principle and research progress., 2020, 18: 5335–5342 (in Chinese with English abstract).
[15] Riedelsheimer C, Czedik-Eysenberg A, Grieder C, Lisec J, Technow F, Sulpice R, Altmann T, Stitt M, Willmitzer L, Melchinger A E. Genomic and metabolic prediction of complex heterotic traits in hybrid maize., 2012, 44: 217–220.
[16] Liu X G, Wang H W, Wang H, Guo Z F, Xu X J, Liu J C, Wang S H, Li W X, Zou C, Prasanna B M, Olsen M S, Huang C L, Xu Y B. Factors affecting genomic selection revealed by empirical evidence in maize., 2018, 6: 341–352.
[17] Wu X, Li Y X, Shi Y S, Song Y C, Zhang D F, Li C H, Buckler E S, Li Y, Zhang Z W, Wang T Y. Joint-linkage mapping and GWAS reveal extensive genetic loci that regulate male inflorescence size in maize., 2016, 14: 1551–1562.
[18] Brown P J, Upadyayula N, Mahone G S, Tian F, Bradbury P J, Myles S, Holland J B, Flint-Garcia S, McMullen M D, Buckler E S, Rocheford T R. Distinct genetic architectures for male and female inflorescence traits of maize., 2011, 7: e1002383.
[19] Song W B, Wang B B, Hauck A L, Dong X M, Li J P, Lai J S. Genetic dissection of maize seedling root system architecture traits using an ultra-high density bin-map and a recombinant inbred line population., 2016, 58: 266–279.
[20] Stuber C W, Edwards M D A, Wendel J F. Molecular marker-facilitated investigations of quantitative trait loci in maize. II: Factors influencing yield and its component traits., 1987, 27: 639–648.
[21] Endelman J B, Atlin G N, Beyene Y, Semagn K, Zhang X C, Sorrells M E, Jannink J L. Optimal design of preliminary yield trials with genome-wide markers., 2014, 54: 48–59.
[22] 李宗泽, 徐晓明, 孙强, 杨彩霞, 许加波, 吴鹏昊. 玉米穗轴长与穗轴粗的QTL定位及全基因组预测. 中国农业大学学报, 2022, 27(4): 44–52.
Li Z Z, Xu X M, Sun Q, Yang C X, Xu J B, Wu P H. QTL mapping and genomic selection of cob length and diameter in maize., 2022, 27(4): 44–52 (in Chinese with English abstract).
[23] Cao S L, Loladze A, Yuan Y B, Wu Y S, Zhang A, Chen J F, Gordon H, Cao J S, Chaikam V, Olsen M, Prasanna B M, San V, Zhang X C. Genome-wide analysis of tar spot complex resistance in maize using genotyping-by-sequencing SNPs and whole-genomeprediction., 2017, 10. doi: 10.3835/plantgenome2016.10.0099.
[24] 贾波, 崔敏, 谢庆春, 严卫古, 印志同. 基于SNP标记的玉米雄穗主要性状QTL定位分析. 西南农业学报, 2019, 32: 1469–1473.
Jia B, Cui M, Xie Q C, Yan W G, Yin Z T. QTL analysis of tassel traits based on SNP markers in maize., 2019, 32: 1469–1473 (in Chinese with English abstract).
[25] Upadyayula N, Da Silva H S, Bohn M O, Rocheford T R. Genetic and QTL analysis of maize tassel and ear inflorescence architecture., 2006, 112: 592–606.
[26] Wang B B, Liu H, Liu Z P, Dong X M, Guo J J, Li W, Chen J, Gao C, Zhu Y B, Zheng X M, Chen Z L, Chen J, Song W B, Hauck A, Lai J S. Identification of minor effect QTLs for plant architecture related traits using super high density genotyping and large recombinant inbred population in maize ()., 2018, 18: 17.
[27] Yang W F, Zheng L Z, He Y, Zhu L Y, Chen X Q, Tao Y S. Fine mapping and candidate gene prediction of a major quantitative trait locus for tassel branch number in maize., 2020, 757: 144928.
[28] Xu G H, Wang X F, Huang C, Xu D Y, Li D, Tian J G, Chen Q Y, Wang C L, Liang Y M, Wu Y Y, Yang X H, Tian F. Complex genetic architecture underlies maize tassel domestication., 2017, 214: 852–864.
[29] Chen Z L, Wang B B, Dong X M, Liu H, Ren L H, Chen J, Hauck A, Song W B, Lai J S. An ultra-high density bin-map for rapid QTL mapping for tassel and ear architecture in a large F2maize population., 2014, 15: 433.
[30] 邵元健. 质量性状和数量性状含义的辨析. 生物学杂志, 2006, (4): 55–57.
Shao Y J. Discrimination of the meaning of qualitative and quantitative traits., 2006, (4): 55–57 (in Chinese).
[31] Cerrudo D, Cao S L, Yuan Y B, Martinez C, Suarez E A, Babu R, Zhang X C, Trachsel S. Genomic selection outperforms marker assisted selection for grain yield and physiological traits in a maize doubled haploid population across water treatments., 2018, 9: 366.
[32] Lorenzana R E, Bernardo R. Accuracy of genotypic value predictions for marker-based selection in biparental plant populations., 2009, 120: 151–161.
[33] Amini F, Franco F R, Hu G P, Wang L Z. The look ahead trace back optimizer for genomic selection under transparent and opaque simulators., 2021, 11: 4124.
[34] Guo R, Dhliwayo T, Mageto E K, Rajas N P, Lee M, Yu D S, Ruan Y Y, Zhang A, Vicente F S, Olsen M, Crossa J, Prasanna B M, Zhang L J, Zhang X C. Genomic prediction of kernel zinc concentration in multiple maize populations using genotyping-by-sequencing and repeat amplification sequencing markers., 2020, 11: 534.
[35] Cao S L, Song J Q, Yuan Y B, Zhang A, Ren J J, Liu Y B, Qu G H, Zhang J G, Wang C P, Cao J S, Olsen M S, Boddupalli P, Vicente F S, Zhang X C. Genomic prediction of resistance to tar spot complex of maize in multiple populations using genotyping-by-sequencing SNPs., 2021, 12: 1438.
[36] Ren J J, Li Z M, Wu P H, Zhang A, Liu Y B, Hu G H, Cao S L,Qu J T, Dhliwayo T, Zheng H J, Olsen M S, Boddupalli P, Vicente F S, Zhang X C. Genetic dissection of quantitative resistance to common rust () in tropical maize (L.) by combined genome-wide association study, linkage mapping, and genomic prediction., 2021, 12: 1338.
[37] Liu X G, Wang H W, Hu X J, Li K, Liu Z F, Wu Y J, Huang C L. Improving genomic selection with quantitative trait loci and nonadditive effects revealed by empirical evidence in maize., 2019, 10: 1129.
QTL locating and genomic selection for tassel-related traits using F2:3lineage haploids
XU Jia-Bo, WU Peng-Hao, HUANG Bo-Wen, CHEN Zhan-Hui, MA Yue-Hong, and REN Jiao-Jiao*
College of Agronomy, Xinjiang Agricultural University, Urumqi 830052, Xinjiang, China
Tassel size affects the nutrient allocation synthesized by photosynthesis in maize, which in turn affects ears development and the yield components, such as kernel rows, kernel number per row, seeding rate, and kernels weight per one hundred. In this study, haploids were induced using the F2:3families, which were constructed from the elite inbred lines Zheng 58 and B73. Genotypes were obtained by 48K liquid phase hybridization capture probes technique, and phenotypes were evaluated in multi-environment trails. QTL mapping was performed using the inclusive composite interval mapping (ICIM) method. The RRBLUP model was used for genomic selection to explore the effects of training population size and the number of SNP markers on prediction accuracy. The results showed that the heritabilities of tassel length, tassel primary branch number, tassel secondary branch number, and tassel branch number were 0.82, 0.88, 0.84, and 0.88, respectively. Two QTL, located in bins 1.03 and 4.09, were detected for tassel length with phenotypic variation explained (PVE) of 6.02% and 11.10%, respectively. Two QTL, located in bins 1.05 and 4.05, were detected for tassel primary branch with PVE of 9.17% and 11.75%, respectively. Two QTL, located in bins 2.03 and 3.06, were detected for the tassel secondary branch with PVE of 5.51% and 5.65%, respectively. Two QTLs, located in bins 1.04 and 4.05, were detected for the tassel branch number with PVE of 9.37% and 10.83%, respectively. Tassel primary branch number and tassel branch number identified a same QTL located in bin 4.05with multiple effects. The prediction accuracy of five-fold cross-validation for genomic selection was 0.36, 0.41, 0.28, and 0.37, respectively. When the training population reached 60% of the total population and the marker density reached 500, a high prediction accuracy could be obtained.
maize (s L.); haploid; tassel; QTLs; genomic selection
10.3724/SP.J.1006.2023.23024
本研究由国家自然科学基金项目(U2003304, 32060484, 32001561), 自治区天山青年优秀青年科技人才培养项目(2018Q019), 新疆自然科学基金项目(2019D01A41), 博士后面上项目(2018M643774), 新疆主要作物生物育种创新工程(一)——玉米生物育种创新工程项目(2021A02001-2)和新疆玉米绿色丰产提质增效技术优化集成及应用项目(2021B02002-2-2)资助。
This study was supported by the National Natural Science Foundation of China (U2003304, 32060484, 32001561), the Scientific and Technological Talent Training Project for Excellent Youth in Tianshan Mountain of Autonomous Region (2018Q019), the Natural Science Foundation of Xinjiang (2019D01A41), the Postdoctoral Project (2018M643774), the Xinjiang Main Crop Biological Breeding Innovation Project (I)—Maize Biological Breeding Innovation Project (2021A02001-2), and the Xinjiang Maize Green High Yield, Quality and Efficiency Technology Optimization Integration and Application (2021B02002-2) Funding.
任姣姣, E-mail: renjiaojiao789@sina.com
E-mail: 1142019044@qq.com
2022-03-03;
2022-06-07;
2022-07-07.
URL: https://kns.cnki.net/kcms/detail/11.1809.S.20220823.1747.002.html
This is an open access article under the CC BY-NC-ND license (http://creativecommons.org/licenses/by-nc-nd/4.0/).
猜你喜欢
语文周报·教研版(2021年28期)2021-08-19
山西农业科学(2020年8期)2020-08-13
陕西农业科学(2019年4期)2019-05-13
河南农业科学(2018年5期)2018-01-19
现代园艺(2017年21期)2018-01-03
新疆农业科技(2016年6期)2016-02-19
中国康复理论与实践(2015年10期)2015-12-24
中国学术期刊文摘(2015年8期)2015-10-29
医学研究杂志(2015年5期)2015-06-10
西北农林科技大学学报(自然科学版)(2015年5期)2015-02-21
