
丝氨酸精氨酸蛋白激酶1在体外促进HepG2细胞干性的获得*
2023-01-15 00:47肖绮雯石永杰周强黄思聪康嘉乐嘉红云彭永正
西部医学 2022年12期
肖绮雯 石永杰 周强 黄思聪 康嘉乐 嘉红云 彭永正
(1. 南方医科大学珠江医院检验医学部,广东 广州 510280;2.广州医科大学附属第二医院检验科,广东 广州 510260;3.南方医科大学珠江医院输血科,广东 广州 510280)
根据GLOBOCAN 2020数据,全球原发性肝癌年新发病数达905,677人,年新增死亡病例830,180人,在全球发病率中排名第五,在男性死亡率中排名第二;肝细胞肝癌(liver hepatocellular carcinoma, LIHC)是原发性肝癌的主要类型[1]。LIHC患者预后差的主要原因是复发率高,且肿瘤干细胞(Cancer stem cells, CSCs)的存在被认为是肝癌复发的根本原因[2]。丝氨酸精氨酸蛋白激酶1(Serine arginine protein kinase 1,SRPK1)是一种mRNA前体的剪接因子,在睾丸癌、乳腺癌、前列腺癌和黑色素瘤中高表达,并促进结肠癌、卵巢癌细胞的药物抗性[3],在非小细胞肺癌中能促进CSCs表型的积累[4]。此前,我们发现高表达SRPK1与肝细胞癌发展相关[5],但其机制尚未完全阐明[6]。本研究中,我们进一步通过调控SRPK1在肝癌细胞株HepG2细胞的表达,研究SRPK1对HepG2细胞肿瘤干性影响,为以SRPK1为靶点的肝细胞癌治疗提供实验依据。
1 材料与方法
1.1 一般材料 HepG2细胞于广州医科大学附属第二医院中心实验室冻存,常规方法复苏冻存细胞,采用体积分数5%胎牛血清、100 mg/L链霉素、100 mg/L青霉素的DMEM培养液,置于37℃、体积分数5% CO2细胞培养箱中常规培养。取对数生长期细胞用于后续实验。
1.2 方法
1.2.1 主要仪器与试剂 胎牛血清、DMEM培养基、胰蛋白酶、嘌呤霉素(美国Gibco公司),表皮生长因子、成纤维细胞生长因子、胰岛素(美国PeproTech公司),TBST缓冲液(美国Thermo Fisher公司),ABI7500 Fast实时荧光定量PCR仪(美国applied biosystems公司),Epics ALTRA流式细胞仪(美国Beckman Coulter公司),桌上离心机(德国sigma公司)。B27、DMEM/F12培养基、总RNA抽提试剂Trizol、Optim-MEM培养基、转染试剂LipofectamineTM2000(美国Invitrogen公司),反转录试剂盒、SYBR Green qPCR Master Mix(加拿大Fermentas公司)。兔抗人SRPK1(美国Abcam公司),兔抗人GAPDH(美国Proteintech公司),HRP-鼠二抗/HRP-兔二抗(美国Merck公司)。牛血清蛋白、荧光染料Hoechst33342、维拉帕米、碘化丙啶(美国Sigma-Aldrich公司)。调节SRPK1表达相关质粒由广州艾基生物技术有限公司提供,重组基因质粒、干扰片段引物设计、合成和测序由广州艾基生物技术完成。
1.2.2 生物信息学分析 从GEPIA2 (http://gepia2.cancer-pku.cn/)获得TCGA中关于LIHC的数据,得到肝癌患者例数为369例,正常对照例数50例。以|Log2FC| Cutoff=0.56,p-value Cutoff=0.01为临界参考,分析SRPK1表达在LIHC组织及正常样本中的差异。分析SRPK1表达在LIHC患者肿瘤分期中的差异性。以肝癌患者肿瘤组织中SRPK1表达的中位数为Cutoff值,将患者分为高、低表达两组,用Kaplan-Meier 法绘制生存曲线,采用 Log-rank 检验比较两组之间的生存差异。运用 TIMER (http://cistrome.org/TIMER/)分析SRPK1表达与LIHC组织中与免疫细胞(B细胞、CD8+T细胞、CD4+T细胞、巨噬细胞、中性粒细胞和树突细胞)浸润丰度的相关性。采用GSEA预测可能受SRPK1调控的信号机制。
1.2.3 过表达SRPK1的HepG2细胞株构建 转染前24 h,在500 μL无双抗完全培养基中接种0.5~2×105个细胞/孔,转染时细胞融合度为80%~90%。按50 μL Opti-MEM稀释0.8 μg质粒(pcDNA3.1-HOMO-SRPK1及pcDNA3.1-HOMO-Vector),50 μL Opti-MEM 稀释2 μL LipofectamineTM2000,混合溶液,轻轻吹吸3~5次混匀,室温静置20 min。转染复合物制备完成后立即加入到24孔细胞板中,100 μL/孔,37℃、5% CO2培养箱中培养6 h左右,换成含5%胎牛血清的培养基,37℃,5%CO2继续培养。如此连续转染3 d,每天上下午各1次,共6次。转染结束后(第4天)用0.5 μg/mL嘌呤霉素筛选细胞,2周后待细胞生长稳定、无明显的死亡细胞时收获细胞。以转染pcDNA3.1-HOMO-SRPK1的细胞株为SRPK1组,pcDNA3.1-HOMO-Vector空白转染细胞为Vector组。
1.2.4 干扰表达SRPK1的HepG2细胞株构建 HOMO-SRPK1-shRNA序列为GAACAACACATTAGCCAACTT。培养皿与先用0.1%明胶铺底,37℃ 孵箱静置 30 mim 后,将细胞数约 3.5×106个的293T细胞铺于含明胶的培养皿上培养24 h后转染。采用PIK 逆转录病毒包装系统。20 μg的pLKO.1-U6-EF1a-copGFP-T2A-puro-SRPK1-shRNA与20 μg包装质粒PIK混匀分别加入2 M CaCl2、380 μL TE缓冲液,充分混匀后,向管子中继续逐滴加入480 μL HEPES缓冲液,将混合液分散滴入培养基中,置于细胞培养箱培养。6 h后用SA缓冲液轻柔洗涤细胞2次,加入含5%FBS的 DMEM培养基。换液后置于细胞培养箱继续培养。转染2~3 d后,每隔6 h收取一次病毒液,储存于-80℃冰箱,连续收取2 d。HepG2细胞铺板步骤同1.2.3,解冻病毒液,向3 mL病毒液中加入 3 mL 5% FBS DMEM培养基、6 μL 聚凝胺(1∶1000),混匀,吸掉细胞培养皿中的培养基,加入混好的病毒液3 mL继续培养4 h,换正常培养基培养2 h,接着继续加入病毒液继续感染细胞,4 h后换正常培养基培养过夜。连续感染3 d,每天上、下午各1次,共6次,筛选细胞步骤同1.2.3。以转染shRNA的细胞株为shRNA组,Scramble空白转染细胞为Scramble组。
1.2.5 Western blot检测各组HepG2细胞SRPK1蛋白水平 用蛋白裂解液裂解转染后的各组细胞,提取总蛋白。BCA法进行蛋白定量,按30 μg/孔蛋白进行10% SDS-PAGE凝胶电泳,湿转PVDF膜后用5%脱脂牛奶室温封闭1 h,一抗(兔抗人SRPK1)4℃封闭过夜,然后用TBST缓冲液洗膜3次,每次10 min,加二抗(1∶2000)室温孵育1 h后,TBST缓冲液洗膜3次,每次10 min,用ECL化学发光试剂曝光。
1.2.6 体外成球实验 配制干细胞培养基50 mL:50x B27 1 mL、100 μg/μL表皮生长因子 10 μL、100 μg/μL成纤维细胞生长因子 10 μL、10% 牛血清蛋白2 mL、1 mg/mL胰岛素 250 uL、DMEM/F12 46.73 mL。SA缓冲液清洗细胞、消化、干细胞培养基垂悬、计数,以约2×103个/孔剂量接种HepG2细胞于低吸附6孔板,每孔约加总量2mL干细胞培养基,第7天计算细胞SFE(SFE=直径大于75 μm的细胞球个数/2000×100%)。
1.2.7 流式细胞术检测SP细胞比例 用胰蛋白酶消化各组HepG2细胞,在含2%胎牛血清的DMEM中以1×106个细胞/mL重悬,然后在37℃预孵育30 min。加入荧光染料Hoechst33342,终浓度为6 μg/mL;其中空白对照组再加入维拉帕米,终浓度50 μmol/mL。37℃避光水浴90 min,每10 min旋转一次。置冰上10 min终止染色,计数细胞;离心后,用4℃含2% 胎牛血清的 PBS洗涤细胞并重悬浮,将分选组调整浓度到1×107/mL以上,用2 μg/mL碘化丙啶来识别死亡细胞,400目纱网过滤细胞。上流式细胞仪对细胞进行分析。Hoechst染料在375nm紫外光激发,450/40带通收集蓝光,695/40带通下收集红光。碘化丙啶在488 nm蓝光处激发,575/26带通下收集红光。以Hoechst染料红光值为X轴,Hoechst染料蓝光值为Y轴作二维散点图,将低Hoechst红及低Hoechst蓝且维拉帕米组缺失的区域设定为SP细胞的阈值,数据采用Summit 5.2软件进行分析。
1.2.8 实时荧光定量PCR法检测干性基因mRNA相对表达量 采用TRizol试剂盒提取各组细胞总RNA,反转录试剂盒反转录合成cDNA,严格按试剂盒说明书进行操作。采用SYBR Green qPCR Master Mix试剂盒进行实时荧光定量PCR,反应体系共10 μL,包括上游引物(20 μmol/L)0.25 μL,下游引物(20 μmol/L)0.25 μL,2×SYBRGreenmix 5 μL,cDNA 0.2 μL,ddH2O 4.3 μL。混匀后瞬时离心,置PCR仪中反应。引物序列见表1。反应条件:95℃ 60 s;94℃ 30 s,50℃30 s,72℃ 45 s,40个循环;72℃10 min。4℃保存。以GAPDH为内参,检测癌细胞干性基因Nanog、Oct4、CD133、Bmi1的mRNA相对表达量,结果以2-△△Ct表示。

表1 实时荧光定量PCR引物序列表Table 1 Primers sequences of Real-time PCR

2 结果
2.1 SRPK1与LIHC疾病进展及免疫细胞浸润丰度的关系 根据从 GEPIA2获得的数据提示,LIHC组织中SRPK1表达明显升高(P<0.01),见图1A。该表达在期癌症的各个分期中也存在差异(F=3.59,P=0.0139),见图1B。高表达SRPK1患者的生存率明显低于低表达患者(HR=1.5,P=0.024),见图1C。在LIHC组织中,SRPK1的表达水平与B细胞(r=0.318,P<0.001)、CD8+T细胞(r=0.159,P=0.003)、CD4+T(r=0.317,P<0.001)、巨噬细胞(r=0.396,P<0.001)、中性粒细胞(r=0.364,P<0.001)和树突细胞(r=0.338,P<0.001)明显相关,见图1D。
2.2 调节HepG2细胞内SRPK1蛋白表达 经广州艾基生物技术公司测序验证,质粒序列与基因库一致。我们构建了SRPK1过表达的稳定株和抑表达稳定株,Western Blot结果所示, SRPK1组SRPK1蛋白相对表达量高于Vector组(t=10.81,P<0.01),shRNA组蛋白相对表达量低于Scramble组(t=10.81,P<0.01),见图2。我们所构建的细胞株能够稳定地过表达或者抑表达SRPK1的表达,可用于后续实验。

图2 调节HepG2细胞SRPK1蛋白表达Figure 2 Regulation of SRPK1 expression in HepG2 cells注:A.重组基因质粒图谱;B.shRNA干扰基因质粒图谱;C.重组基因、shRNA测序验证;D.WB验证HepG2细胞SRPK1蛋白表达。①P<0.01
2.3 SRPK1对HepG2细胞干性的影响 SRPK1组SFE高于Vector组(t=9.35,P<0.01),shRNA组低于Scramble组(t=3.01,P=0.04),见图3A。SRPK1组SP细胞比例高于Vector组(t=7.72,P<0.01),shRNA组SP细胞比例低于Scramble组(t=5.22,P<0.01),见图3B。体外成球能力和SP细胞比例是肿瘤细胞干性表型的重要体现。结果揭示SRPK1参与HepG2细胞肿瘤干性调节,过表达SRPK1基因显著提高了HepG2细胞的干性特征。

图3 SRPK1对HepG2细胞干性的影响Figure 3 The effect of SRPK1 on tumor stemness in HepG2 cells注:A.各组细胞体外成球能力;B.各组SP细胞比例。①P<0.01,② P<0.05
2.4 SRPK1对HepG2细胞干性相关基因的影响 SRPK1组Nanog、Oct4、CD133、Bmi1 mRNA相对表达量高于Vector组(P<0.05),Scramble组高于shRNA组(P<0.05),见图4。结果表明HepG2细胞的4个干性标志基因受SRPK1表达调节,其中变化最大的基因为Bmi1。
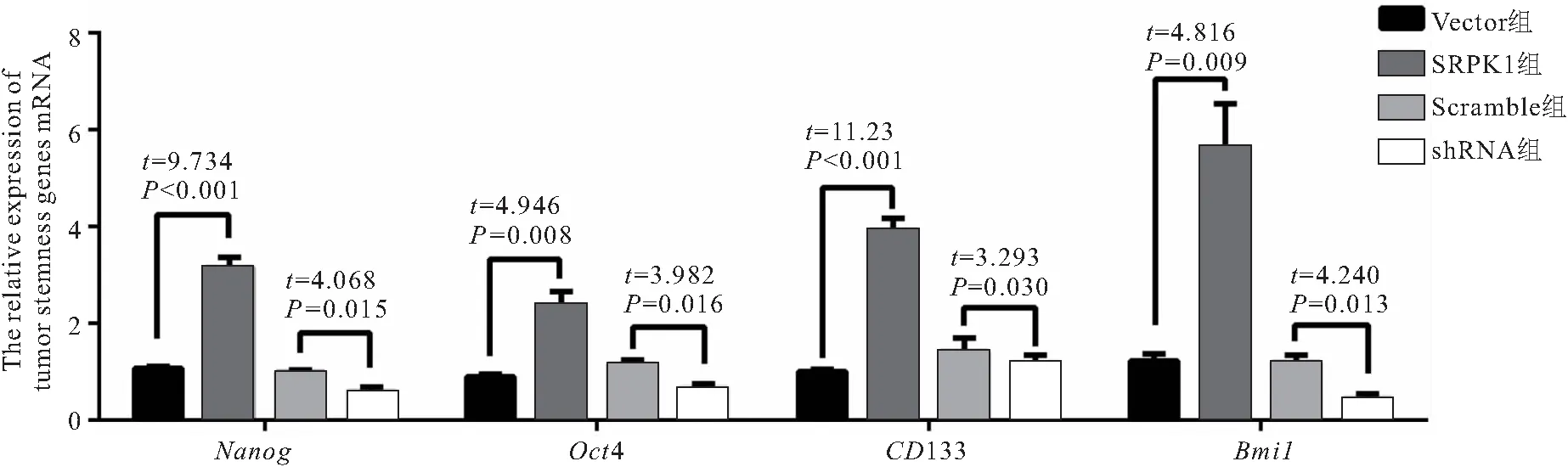
图4 SRPK1对HepG2细胞干性基因的影响Figure 4 The effect of SRPK1 on tumor stemness relative genes in HepG2 cells
2.5 SRPK1可能促进HepG2细胞Wnt/β-catenin信号通路活化 目前,有报道称Wnt/β-catenin通路是诱导CSC表型的积累的关键途径。用GSEA软件分析了肝癌数据库中SRPK1的表达可能调控的信号机制,结果发现,SRPK1表达在Wnt/β-catenin通路活化的样本中高富集(P<0.05),见图5。

图5 SRPK1与Wnt/β-catenin通路的相关性Figure 5 Correlation of SRPK1 and Wnt/β-catenin pathway activation
3 讨论
LIHC仍然是目前一个严重的世界健康问题,高复发率是其治疗的重大挑战[7];新的治疗靶点和标志物一直在不停研发当中[8-9]。目前学界对于促癌基因推动细胞癌变的作用主要集中研究6种能力:CSCs表型的积累,细胞增殖,化疗敏感性,逃避程序性死亡,组织侵袭和转移,以及持续的血管生成[6]。CSCs是存在于肿瘤组织中的一小部分细胞亚群,具有自我更新以及多向分化潜能等干细胞特性。SP细胞可以有效地作为鉴别肝癌CSCs的标志物,并通过上调干性基因和肿瘤形成来启动肿瘤发生,肝癌细胞中SP细胞越多,其转移能力和肿瘤形成能力越强[10-12]。肝CSCs促进肝细胞癌高侵袭性、耐药、易复发、易转移、预后差等恶性特点[13],因此,直接靶向肝CSCs被认为是改善LIHC患者预后的一种有效的治疗策略[2,14-15]。SRPK1是剪接因子蛋白激酶家族成员之一,是一类保守的真核激酶,能使丝氨酸/精氨酸蛋白磷酸化,刺激mRNA前体的可变剪接加工过程,并通过调控哺乳动物胚胎干细胞的泛素信号通路,确保了神经发育基因表达的正确调控[16]。调节SRPK1表达可通过NF-kappaB通路有效改善结肠癌的化疗耐药[17],以SRPK1作为靶点的方案被证明对白血病治疗有效[18],特异性抑制SRPK1抗癌药物的开发已取得一定进展[19]。在既往对SRPK1促瘤能力的研究中发现,SRPK1可促进肝癌细胞的增殖[20]。本研究通过对TCGA数据库进行分析,结果显示SRPK1在LIHC组织中表达显著高于正常组织,高表达SRPK1患者总生存率显著低于低表达患者。同时,通过TIMER数据库分析发现SRPK1表达与LIHC组织中多种免疫细胞浸润正相关。LIHC组织中肿瘤相关免疫细胞通常呈高浸润状态,且浸润水平与多项临床病理特征和预后密切相关[21]。结果再次证明SRPK1在HCC中与肿瘤发展及患者的不良预后高度相关[5]。
既往研究报道,SRPK1能促进非小细胞肺癌肿瘤干细胞表型的获得[4],也是多种消化系统肿瘤的致癌基因和潜在的治疗靶点,但SRPK1对肝癌细胞干性表型的影响目前缺乏报道[6]。为了探讨SRPK1对肝癌细胞肿瘤干性的影响,课题组通过前期研究[5]选取中等表达SRPK1的HepG2细胞,构建了不同SRPK1表达的细胞模型。通过一系列体外细胞实验证明,过表达SRPK1明显增强HepG2细胞在体外的成球能力,也增加了癌细胞群中SP+细胞的比例,而在抑制SRPK1表达后则观察到相反的实验结果。Nanog和Oct4是干细胞自我更新及保持未分化特性的关键因子,具有使终端分化细胞重新进入细胞周期进行增殖的能力[22]。CD133高表达可明显提高肿瘤细胞的成球能力,并诱导癌细胞发生免疫逃逸和对抗程序性凋亡而获得更明显的干细胞表型[23]。Bmi1基因参与肝癌细胞干性功能的维持[24]。我们发现过表达SRPK1会促使HepG2细胞Nango、Oct4、CD133以及Bmi1的表达增高,而抑制SRPK1表达后,4个肿瘤干性基因表达相应降低。
Wnt/β-catenin通路的过度活化可以直接促进肿瘤的干性获得、耐药及免疫逃逸[25],Gong等[4]对非小细胞肺癌研究指出,Wnt/β-catenin通路是SRPK1诱导CSC表型的积累的关键途径。本次GSEA分析显示SRPK1高表达与Wnt/β-catenin通路活化呈正相关,预测SRPK1在体外促进HepG2细胞干性的获得可能与Wnt/β-catenin通路的活化有关。
综上所述,SRPK1在体外可促进HepG2细胞肿瘤干性的获得,结论与SRPK1对其他类型肿瘤(肺癌、前列腺癌、结肠癌等)的作用一致,机制可能与Wnt/β-catenin通路活化有关,研究结果为以SRPK1作为肝细胞肝癌的治疗靶点提供了新的理论依据。但本研究具有一定局限性,首先流式细胞实验结果显示HepG2细胞自身SP亚群所占比例较低,因此以SRPK1为治疗靶点抑制干性作为LIHC的治疗方向仍然待进一步论证,我们计划在往后研究中以SP亚群比例更高的细胞进行实验,并结合裸鼠体内成瘤实验进行验证,增加本研究结论的说服力。其次,本次研究未通过相关实验确证Wnt/β-catenin通路在SRPK1促进HepG2细胞干性获得过程中的作用,我们下一步将针对该通路的作用靶点进行更具体的分子机制分析。
4 结论
SRPK1与LIHC发展紧密联系,其高表达既预示肿瘤的不良预后,也促进肝癌细胞干性表型的获得,机制可能通过与Wnt/β-catenin通路激活相关。
猜你喜欢
世界科学技术-中医药现代化(2021年5期)2021-11-05
中国科技纵横(2021年24期)2021-03-02
蚕桑通报(2020年1期)2020-07-10
中国土壤与肥料(2018年5期)2018-11-05
中成药(2018年6期)2018-07-11
现代园艺(2017年21期)2018-01-03
方圆(2017年12期)2017-07-17
中国康复理论与实践(2015年10期)2015-12-24
家庭医学(2015年8期)2015-09-10
医学研究杂志(2015年5期)2015-06-10
