
氮化碳与介孔氧化硅协同催化CO2环加成研究
2023-01-07 12:43王忠华黄春霞邵守言符微微
安徽化工 2022年6期
王忠华,唐 丽,黄春霞,邵守言,符微微,吴 杰
(1.江苏索普(集团)有限公司,江苏 镇江 212006;2.中国科学院化学研究所,北京 100190)
碳排放引起的温室效应威胁着人类的生存与发展。为此,中国制定了2030年碳达峰、2060年碳中和的碳减排目标。清洁能源取代化石能源是实现碳减排最有效的手段,然而,在很多重要领域,化石能源不可被替代,由此产生的CO2可通过碳捕捉与封存进行处理,但该技术成本高昂[1]。CO2的资源化是指在催化剂作用下,将CO2催化活化,转化为有价值的化合物,变废为宝,实现碳减排[2]。然而,CO2分子能量低,目前广泛研究的光、电催化CO2还原需要断开CO键,能耗大[3]。CO2与环氧化合物环加成不需要断开CO键,能耗低,是更为可行的CO2资源化利用方法[4]。产物环状碳酸酯是高沸点的极性化合物,可用作燃料添加剂、非质子溶剂、电池电解质和聚碳酸酯单体等,具有很高的价值[5]。
石墨相氮化碳(CN)是一种富含氮功能基团的层状高分子,被广泛用于催化CO2与环氧化合物环加成[6]。然而,体相CN的活性低,这是由于CN表面功能基团少,比表面积低。为此,研究者通过杂原子修饰[7],加入第二组份等多种方法引入活性功能基团[8],提高了CN的催化活性。增加比表面积是提高CN活性的另一种方法,比如,将CN负载到介孔分子筛SBA-15中,实现了催化活性的大幅提高[9]。然而,SBA-15尺寸大,在微米级,反应物的扩散距离长,很难到达孔道内部,影响了CN的催化性能。更重要的是,研究者没有对催化机理进行深入研究,仅将活性提高的原因归结为负载后的CN表面活性位点增多。
基于此,本文将CN负载到尺寸较小的介孔氧化硅纳米球(MSN)的孔道中,减小了反应物的扩散距离,提高了催化活性。另外,考虑到CN中的含氮功能基团能高效活化CO2,但对环氧的活化性能较低,不能解释CN/MSN的极高催化活性。为此,进一步研究了MSN中硅羟基与CN的协同催化作用,这为高性能CN催化剂的设计提供了新思路。
1 实验部分
1.1 试剂和仪器
二聚氰胺、环氧氯丙烷、环己烷均为分析纯,国药试剂;正硅酸乙酯(TEOS)、十六烷基三甲基氯化铵(CTAC)、三乙醇胺(TEA)、ZnBr2和活性炭(AC),均为分析纯,阿拉丁试剂公司;CO2、Ar、高纯气;环氧氯丙烷使用前用分子筛脱水,其余试剂使用前未经处理。
GC-2014型气相色谱仪;SEM-4800型扫描电镜;JEM-2011型透射电镜;康塔Autosorb-iQ-3型比表面物理吸附分析仪;D/max 2500型X射线粉末衍射仪;TENSOR-27型傅里叶红外光谱仪;ESCALAB220I-XL型X射线光电子能谱仪。
1.2 催化剂制备
将24 mL的CTAC水溶液(25%)加入到100 mL单口瓶中,加入0.18 g的TEA和36 mL的二次水,60℃下搅拌30 min后加入TEOS的环己烷溶液(20 V/V %)20 mL,慢速搅拌24 h。离心分离产物,用乙醇洗涤3次,110℃干燥6 h。将所得白色产物置于马弗炉中,550℃焙烧2 h,去除模板,得到MSN。
将0.2 g二聚氰胺加入到6 mL二次水中,60℃搅拌10 min。完全溶解后,加入1.0 g的MSN,超声分散,用旋转蒸发仪60℃干燥。将所得固体置于管式炉中,Ar保护下,以3℃·min-1的速率升温至550℃,保温2 h,得到淡黄色的产物CN/MSN。
1.3 CO2与环氧氯丙烷环加成反应
环加成反应在100 mL高压反应釜中进行。将50 mg催化剂、0.288 g的ZnBr2(1 mol%)和10 mL环氧氯丙烷加入到反应釜中,超声分散10 min。用Ar置换3次,升温至140℃后,充入3.5 MPa CO2。反应3 h后,用冰水浴冷却反应釜、泄压。将催化剂离心分离,用气相色谱分析上层清液。
2 结果与讨论
载体MSN通过文献方法合成[10]。如图1a的SEM图所示,MSN为纳米球状,尺寸较为均一,平均直径为124 nm。较小的粒径缩短了反应物的扩散距离,可降低扩散的影响。浸渍前驱体二聚氰胺,高温聚合后,得到负载型CN/MSN。通过重量变化计算得出的负载量为8.6 wt%。负载后,MSN的形貌没有发生明显变化(图1b)。图1c、d分别是MSN和CN/MSN的TEM图。MSN具有放射型的介孔结构,孔径较为单一,尺寸在5 nm左右。规则的孔结构有利于反应物的扩散。CN/MSN保留了MSN的放射型介孔结构,但孔道变得模糊,说明二聚氰胺在MSN孔道内部限域聚合。限域作用使生成的CN以纳米态负载在MSN孔道的内部[11]。

图1 MSN(a)和CN/MSN(b)的SEM图;MSN(c)和CN/MSN(d)的TEM图Fig.1 SEM images of MSN(a)and CN/MSN(b);TEM images of MSN(c)and CN/MSN(d)
N2吸附-脱附等温线如图2a所示。通过计算得出,MSN和CN/MSN的BET比表面积分别为684.5 m2·g-1和621.3 m2·g-1。负载后,比表面积降低了约9%。MSN是典型的IV型吸附-脱附等温线,P/P0在0.4~0.8范围内明显的H1滞后环表明介孔的存在[12]。与MSN相比,CN/MSN的吸附-脱附曲线没有发生明显变化,但滞后环略微提前,说明孔径变小。为此,对孔径分布进行了分析。图2b的BJH脱附孔径分布表明,MSN具有介孔结构,孔径分布在3~9 nm,平均孔径6.1 nm。负载CN后,孔径分布变化不大,平均孔径减小至5.8 nm。虽然CN在孔道的负载降低了比表面积,减小了孔径,但CN/MSN仍然具有很高的比表面积、适中的孔径大小和较均一的孔径分布,说明在高温制备条件下,CN和SiO2具有较强的亲和性。纳米态的CN生长在MSN的孔壁,具有极好的分散性,没有发生团聚和堵塞MSN的孔道。
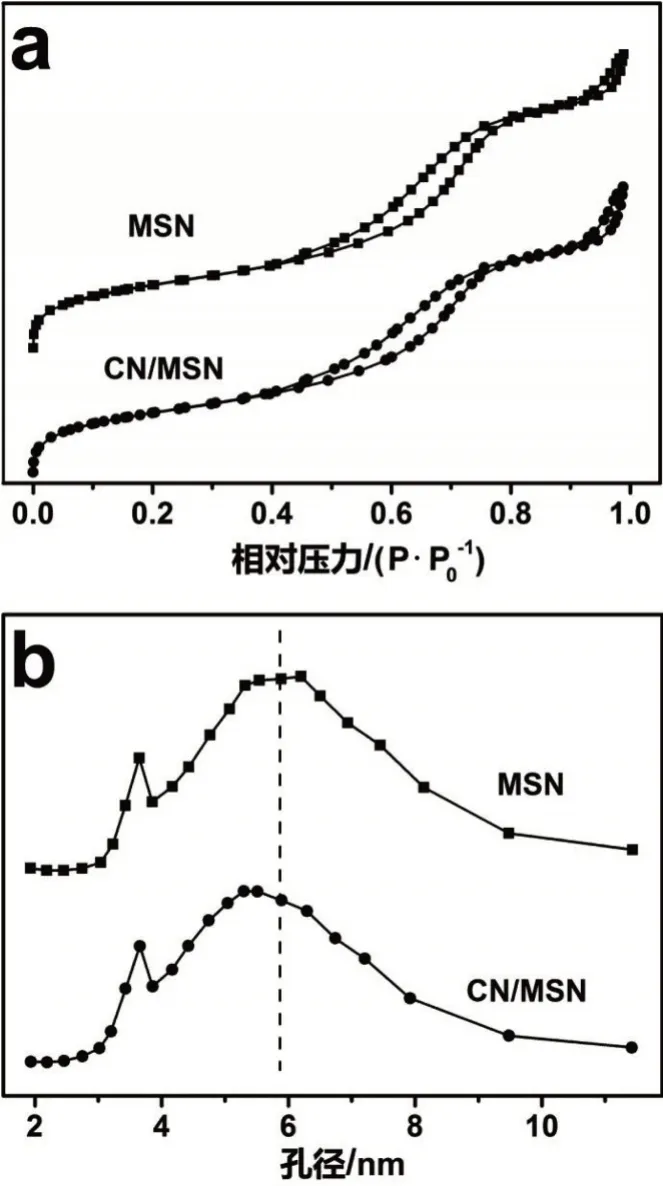
图2 MSN和CN/MSN的N2吸附-脱附等温线和BJH孔径分布Fig.2 N2 adsorption-desorption isotherms(a)and BJH pore distributions(b)of MSN and CN/MSN
XRD用于表征材料的晶相结构。如图3所示,体相CN在2θ为13.1°和27.3°处有明显的衍射峰,分别对应重复的三-S-三嗪单元和层层堆积的二维结构[13]。MSN只有一个在2θ为15°~30°间的馒头峰,说明MSN为无定型态的SiO2。负载CN后的CN/MSN没有发现CN的特征衍射峰。一方面,CN的负载量较低(8.6 wt%),衍射峰较弱;另一方面,由于MSN孔道的限域作用,CN以纳米态分散在MSN的孔道内,晶粒小,衍射信号弱,这与TEM和N2吸附-脱附结果一致。纳米态的CN含有更多的结构缺陷和功能基团,这些缺陷和功能基团往往是高活性的催化位点。
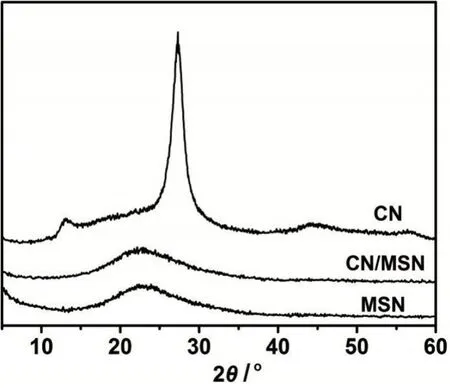
图3 CN、MSN和CN/MSN的X射线粉末衍射图Fig.3 Powder XRD patterns of CN,MSN and CN/MSN
FT-IR用于表征材料的功能基团。由图4可见,CN在798 cm-1处的吸收来自于三嗪杂环;1 200~1 650 cm-1范围内的谱带归属于CN的伸缩振动和NH的弯曲振动;3 183 cm-1附近的吸收归因于NHx。该功能基团是不完全聚合产生的终端基团。对于MSN,955 cm-1处的吸收对应的是SiOH的弯曲振动。3 000~3 600 cm-1范围内的宽峰说明MSN含有大量的羟基。负载CN后,CN/MSN同时显示了CN和MSN的IR吸收,说明CN被成功负载到MSN中。此外,在2 225 cm-1处发现一较强的新信号,对应的是C≡N基团[14]。MSN孔道的限域作用使二聚氰胺聚合不完全,产生了大量的NHx和C≡N等终端基团。这些终端功能基团是催化CO2活化的高活性位点。
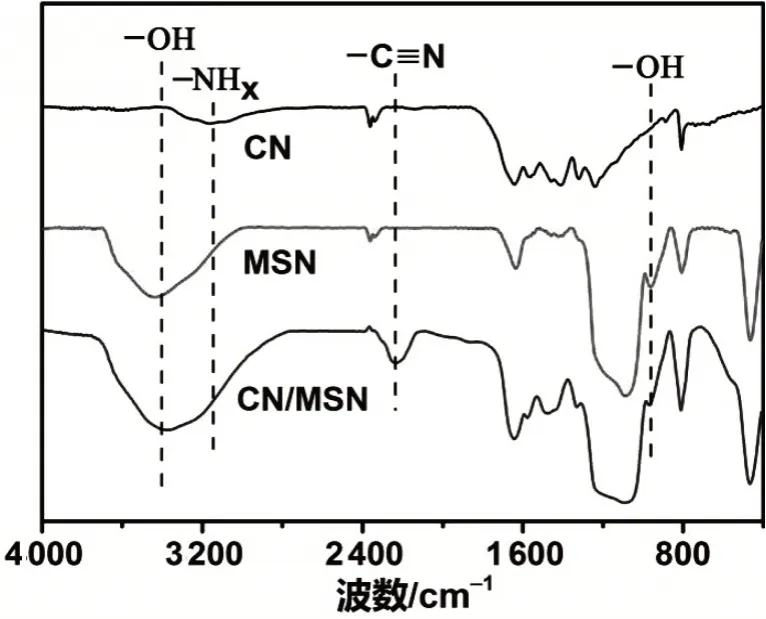
图4 CN、MSN和CN/MSN的红外光谱图Fig.4 FT-IR spectra of CN,MSN and CN/MSN
用XPS对材料表面化学和功能基团作进一步表征。如图5a所示,CN的C 1s峰可拟合为两个峰,284.8 eV处对应的是石墨或无定形碳,来自于污染碳或CN的缺陷位点,该峰较弱,说明体相CN的缺陷含量低。CN中的CN杂环显示在288.2 eV处,该峰较强,说明体相CN具有较完整的三-S-三嗪杂环结构。对于CN/MSN,该峰强度显著下降,表明大量三-S-三嗪杂环结构被破坏;另外,其峰位向高结合能方向移动,进一步表明其结构被破坏,导致C的化学环境发生变化。含N功能基团是CN活化CO2的活性位点[15]。因此,对N 1s进行了分峰处理。图5b中,CN在398.6 eV处的峰对应的是本体三-S-三嗪杂环。该峰强度大,与C 1s的结果一致。桥联N显示在399.4 eV,同时,400.8 eV处有一较小峰,归属于终端NHx基团,这与FT-IR的结果一致。该峰所占比例较小,说明终端NHx基团含量较低。对于CN/MCN,三-S-三嗪杂环的信号大幅下降,表明CN的主体结构大量被破坏,与C 1s的结果一致。终端NHx所占比例大幅提高,表明CN/MCN含有大量的缺陷和终端功能基团[16]。

图5 CN和CN/MSN的C 1s(a)和N 1s(b)XPS谱图Fig.5 C 1s(a)and N 1s(b)XPS spectra of CN and CN/MSN
以环氧氯丙烷为模型底物,考查CN/MSN催化CO2和环氧化合物环加成制备环状碳酸酯的性能。如图6所示,MSN的活性极低,而CN有一定的催化性能,环氧氯丙烷的转化率为9.7%。这是由于CN含有少量的NHx功能基团,可催化CO2和环氧的加成反应[17]。将CN负载到MSN上后,所得CN/MSN的催化性能大幅提高,环氧氯丙烷的转化率达到95.4%,与体相CN相比,提高了近10倍。需要注意的是,CN/MSN中,CN的负载量是8.6wt%,按CN计算,活性提高了约110倍。CN/MSN大的比表面积、发达的孔结构和较小的粒径都有利于提高其催化性能。然而,研究指出,CN中的含N基团能高效活化CO2,但对环氧的催化活化性能较差[18]。CN/MSN极高的催化活性说明CN/MSN中可能存在可高效活化环氧的活性位点。
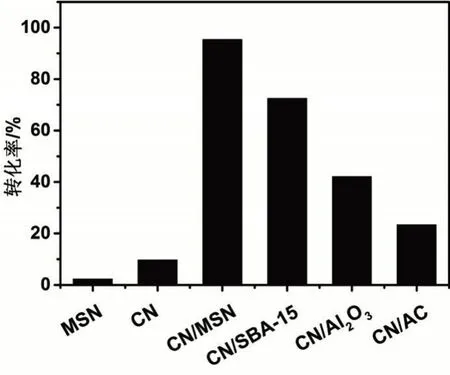
图6 不同催化剂的性能Fig.6 Performance of different catalysts
XRD和FT-IR结果表明,MSN由无定型的SiO2组成,含有大量的硅羟基。此外,MSN比表面积大,孔壁薄,硅羟基暴露在表面,极易和反应物接触。同时,大量研究表明,羟基是活化环氧的活性位点[19]。因此,CN和硅羟基的协同催化作用可能是CN/MSN高活性的原因。为了证明该假设,分别以SBA-15和活性炭(AC)为载体,合成了CN/SBA-15和CN/AC两种催化剂。SBA-15具有与MSN相同的化学组成,含有大量的硅羟基。以CN/SBA-15为催化剂时,环氧氯丙烷的转化率为72.5%,略低于CN/MSN。这可能是由于SBA-1的粒径较大,反应物很难扩散到孔道内部,降低了催化活性。以CN/AC为催化剂时,环氧氯丙烷的转化率进一步下降为23.4%。AC的比表面积为1 800 m2·g-1,远高于MSN和SBA-15。然而,AC不含羟基。因此,CN/AC对环氧的活化性能低,导致催化CO2与环氧环加成的活性低。
基于以上实验数据,结合前人的研究成果[20],我们提出了图7的CN和硅羟基协同催化作用机理:首先,硅羟基与环氧形成氢键,富含胺基的CN与CO2成环,环氧和CO2被活化。接着,活化后的CO2进攻被活化的环氧中的C原子,发生开环。最后,CO2与环氧成环,生成环状碳酸酯。同时,催化剂被还原为初始态,进行下一轮催化反应。

图7 可能的反应机理Fig.7 Possible reaction mechanism
3 结论
采用浸渍前驱体、高温焙烧法制备了CN/MSN,CN的含量为8.6wt%。在140℃,3.5 MPa的CO2,50 mg的CN/MSN,1 mol%的ZnBr2和10 mL环氧氯丙烷的条件下反应3 h,环氧氯丙烷的转化率达到95.4%,远高于体相CN的9.7%,同时揭示了CN与硅羟基的协同催化机理。
猜你喜欢
大电机技术(2022年5期)2022-11-17
内燃机与动力装置(2022年1期)2022-03-21
哈尔滨工程大学学报(2021年2期)2021-03-19
湖北农机化(2020年4期)2020-07-24
世界农药(2019年4期)2019-12-30
新课程·下旬(2019年7期)2019-09-17
发明与创新·中学生(2017年11期)2017-12-07
筑路机械与施工机械化(2017年5期)2017-08-31
筑路机械与施工机械化(2017年6期)2017-07-10
航天制造技术(2016年6期)2016-05-09
