
聚乙烯醇/海藻酸钠复合水凝胶的制备及吸附性能研究
2023-01-07 12:43杨青峰史然邢宏龙
安徽化工 2022年6期
杨青峰,史然,邢宏龙
(安徽理工大学 化学工程学院,安徽 淮南 232001)
水凝胶是一类以化学、物理等方式交联的具有三维网络结构的聚合物[1-2],具有众多的工业应用,例如,水凝胶可用于伤口敷料[3]、智能传感器[4]、药物释放[5]、组织工程等[6]。聚乙烯醇(PVA)是一种常见的线性聚羟基聚合物,具有较高的成膜能力和化学稳定性,其无毒、可生物降解,并且是水溶性的[7],具有制备理想“绿色”水凝胶的先天优势[8]。但单一的PVA水凝胶易发生溶胀和粘连,稳定性较差[9]。海藻酸钠(SA)是一种从天然海藻中提取或由细菌产生的水溶性阴离子生物聚合物[10]。因其富含羟基、羧基,与染料及金属离子亲和力强[11],而单组分SA水凝胶存在机械性能较差、凝胶成型性差、吸附稳定性不好等问题,严重限制了其广泛应用[12-13]。
PVA/SA复合水凝胶能结合PVA和SA的优点,解决单一组分水凝胶材料机械性能或稳定性较差的问题。但是,研究工作的难点在于控制原料的质量比以构建互穿网络结构,提高水凝胶的吸附性能。本文通过冻融方法向PVA水凝胶中引入海藻酸钠,以实现PVA与SA在性能上的优势互补。
1 实验部分
1.1 原料及仪器
聚乙烯醇(1799),阿拉丁化学试剂有限公司;海藻酸钠、氯化铁,国药集团化学试剂有限公司;无水氯化钙,科隆化学试剂厂;磺基水杨酸,天津永大化学试剂有限公司。
TGL-18C型高速台式离心机,上海安亭科学仪器厂;Nicolet is50型FT-IR红外光谱仪,美国赛默飞世尔科技公司;S4800型场发射扫描电子显微镜,美国FEI公司;UV-2550型紫外分光光度计,日本岛津公司;冷冻干燥机,河南信陵仪器设备有限公司;力显拉伸机,东莞市力显仪器科技有限公司。
1.2 PVA/SA复合水凝胶的制备
PVA/SA复合水凝胶是通过冻融[14]方法制备的。首先,将10.00 g的PVA颗粒溶解在90 mL的去离子水中,在95℃下油浴1 h,制备10wt%的PVA溶液,之后将溶液冷却到室温。然后用97 mL去离子水溶解3.00 g SA粉末,于80℃搅拌3 h至充分溶解,得到3wt%的SA溶液,之后将溶液冷却到室温。将上述PVA溶液和SA溶液按4∶1,3∶2,1∶1,2∶3,1∶4的不同质量比混合1 h,得到均匀的混合溶液。将混合溶液消泡后注入由一对玻璃板组成的自制模具中,在冰箱中以-18℃的温度冷冻4 h,然后从冰箱取出试样,常温下解冻2 h。该冷冻与解冻过程反复进行3次得到PVA/SA复合水凝胶。在0.05 mol/L的氯化钙溶液中浸泡12 h进行交联后,取出复合水凝胶,用去离子水将表面多余的氯化钙溶液洗掉。交联后的PVA/SA复合水凝胶在去离子水中溶胀至平衡,并在50℃下干燥至不再有重量变化。
1.3 PVA/SA复合水凝胶的吸附性能
将烘干的水凝胶吸附剂加入Fe3+溶液中,直到吸附达到平衡。用高速离心机将溶液离心,取上清液适当稀释后用紫外分光光度计在510 nm处[15]测量吸光度。吸附平衡时的吸附量Qe(mg/g)、时间t的吸附量Qt(mg/g)和吸附率R(%)分别按以下公式计算[16]:


式中:Qe—平衡吸附量,mg/g;Qt—t时刻的吸附量,mg/g;Co—初始浓度,mg/L;Ce—平衡时溶液浓度,mg/L;Ct—吸附t时刻浓度,mg/L;V—溶液体积,L;m—添加水凝胶的质量,g;R—吸附率,%。
2 结果与讨论
2.1 扫描电子显微镜(SEM)分析
图1显示了不同质量比复合水凝胶的SEM图像。从图1(a)可以看出,纯PVA水凝胶的表面均匀,没有明显的孔隙结构。随着SA含量的增加,样品表面变为网状和多孔,如图1(b,c)所示,表明样品表面的聚合物网络变得松散而不是致密。然而,当SA含量达到50%时,表面显示出斑点和不均匀,如图1(d,e,f)所示。

图1 PVA/SA复合水凝胶的SEM图像
2.2 傅里叶变换红外光谱(FTIR)分析
通过FTIR对复合水凝胶的基团变化进行分析研究。图2是PVA、SA、PVA∶SA(1∶1)水凝胶的FTIR图谱。如图2所示,从PVA的FTIR光谱中可以看出,O-H和C-H的拉伸振动分别在3 307 cm-1和2 942 cm-1处产生吸收峰,而C-H的弯曲振动峰在1 423 cm-1处,1 092 cm-1处产生的吸收峰是由C-O伸缩振动引起的特征峰;SA的FTIR显示,O-H伸缩振动导致3 271 cm-1的吸收峰,SA大分子中C-H伸缩振动导致2 938 cm-1的吸收峰,-COO-的不对称和对称伸缩振动是分别导致1 593 cm-1和1 416 cm-1的吸收峰[17],C-O伸缩振动吸收峰位于1 030cm-1。SA的加入导致了O-H拉伸振动吸收峰向高波数移动,说明SA的存在削弱了PVA分子之间的氢键作用。

图2 PVA、SA和PVA/SA复合水凝胶的FTIR图谱
2.3 PVA/SA质量比对吸附性能的影响
如图3所示,很明显可以看出随着SA含量的增加,对Fe3+的吸附率逐渐增大。这是因为随着SA的增多,复合水凝胶上的-COOH和-OH官能团数目逐渐增多,有利于促进对Fe3+的吸附。因此选择PVA∶SA质量比为1∶4的复合水凝胶样品做进一步的研究。

图3 PVA/SA质量比对吸附性能的影响
2.4 Fe3+初始浓度对吸附性能的影响
如图4所示,Fe3+初始浓度越低,加入等量吸附剂时水凝胶对Fe3+的吸附效果越明显。这一结果可能是由于当Fe3+的初始浓度较低时,氯化铁溶液中的铁离子较少,PVA/SA复合水凝胶表面有大量的吸附位点[18],可以与Fe3+迅速结合,从而有效地进行吸附。然而,当Fe3+溶液的初始浓度增长时,溶液中的Fe3+数量也会增加。此外,水凝胶表面有一定数量的吸附点对Fe3+的吸附是无效的,这降低了Fe3+的吸附率。

图4 Fe3+初始浓度对吸附性能的影响
2.5 pH值对吸附性能的影响
如图5所示,Fe3+的吸附量及吸附率均随pH的上升呈增加的趋势,pH由4到5时,吸附量增加缓慢,几乎达到平衡。当pH值较低时,溶液中就有更多的H+,PVA/SA水凝胶中的羧酸根质子化程度相对较大(-COO-到-COOH)[19],这导致水凝胶网络上的吸附位点数量减少,同时,过多的H+增加了H+和Fe3+之间的静电排斥,降低了水凝胶和Fe3+之间的静电作用。因此,pH值减弱了对Fe(III)的吸附作用。伴随着pH值的增加,羧基发生电离,增加的-COO-使水凝胶带负电,Fe(III)与-COO-发生配位反应,水凝胶与Fe3+之间的离子交换、静电作用增强,导致吸附量增加。

图5 pH值对吸附性能的影响
2.6 水凝胶用量对吸附性能的影响
如图6所示,Fe3+的吸附受到水凝胶上的吸附活性点的限制。低水凝胶用量容易达到吸附饱和。随着水凝胶用量增大,为Fe3+提供更多的吸附位点,大量的活性位点处于非饱和吸附状态。由于Fe3+溶液的浓度一定,分配到复合水凝胶每个单位吸附位点的Fe3+量随着水凝胶用量的增加而减少,所以吸附量开始减少。

图6 水凝胶用量对吸附性能的影响
2.7 吸附时间对吸附性能的影响
如图7所示。水凝胶对Fe3+的吸附量随着吸附时间的增加而逐渐增加,其中水凝胶在前90 min内呈现出快速吸附模式。这可能是因为在初始阶段,水凝胶中存在很多活跃的吸附位点,导致吸附驱动力大,Fe3+与吸附位点的结合速率快,溶液开始渗透到水凝胶基体中,促进了Fe3+的吸附。90 min后,吸附量的增长放缓,这是因为水凝胶内部的大部分吸附位点已经被Fe3+占据,使得Fe3+进入水凝胶网络内部的驱动力减弱,所以吸附量逐渐减少。150 min时,Fe3+吸附达平衡状态,最后平衡吸附量达83.11 mg/g。
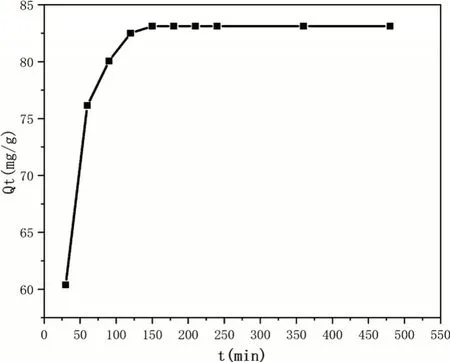
图7 吸附时间对吸附性能的影响
2.8 吸附动力学分析
使用准一级、准二级动力学模型分析水凝胶的吸附过程,分别代表物理吸附、化学吸附[20]。由以下公式表示:
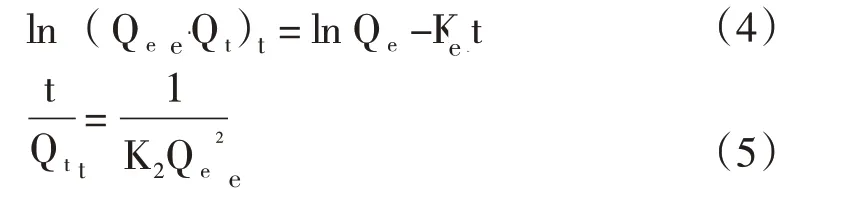
式中,Qe—吸附Fe3+平衡时的吸附量,mg/g;Qt—t时的吸附量,mg/g;K1—准一级动力学吸附速率常数,min-1;K2—准二级动力学吸附速率常数,g/(mg·min);t—时间,min。
“速率控制”经常使用颗粒内扩散模型进行预测,其模型方程通常为[21]:

式中,Qt—t时的吸附量,mg/g;Kp—颗粒内扩散常数,mg/(g·min1/2);C—与边界厚度有关的常数;t—吸附时间,min。
三个动力学模型的拟合结果见图8,拟合参数见表1。
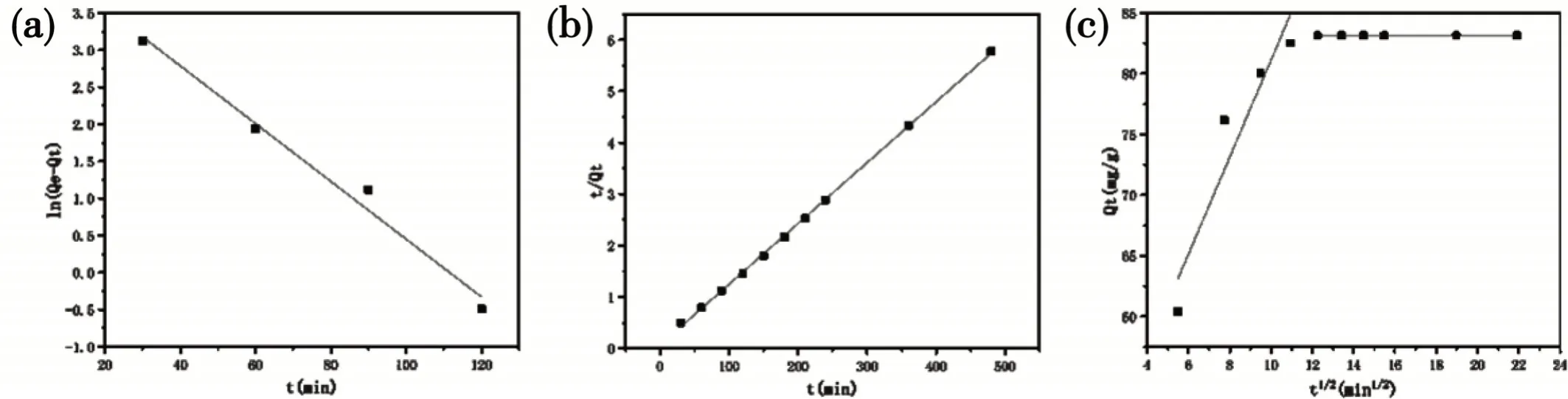
图8 (a)准一级动力学模型的拟合曲线;(b)准二级动力学模型的拟合曲线;(c)颗粒内扩散模型的拟合曲线

表1 动力学参数
如表1所示,水凝胶的准二级动力学模型比准一级动力学模型具有更强的相关系数,表明前者更适合描述吸附过程[22]。准二级动力学模型准确地描述了PVA/SA水凝胶的吸附机制,其主要受化学吸附控制。
图8(c)是PVA/SA水凝胶的粒内扩散模型的拟合结果,从图中可以看出,在整个吸附时间内,拟合的曲线不是线性的,拟合曲线与原点没有交点,表明除了颗粒内扩散外,它还受到边界层扩散的影响[23]。粒子内扩散系数Kp下降,而扩散边界层C增加。这一现象表明,当PVA/SA刚开始吸附Fe3+时,扩散边界层较薄,扩散阻力较小。随着Fe3+在PVA/SA水凝胶上占据的吸附位点逐渐增加,扩散边界层增加,扩散阻力相应增加,导致扩散速率变慢。
3 结论
以具有良好生物相容性和生物降解性的材料PVA和具有低膨胀性的天然多糖材料SA为基础,采用冻融法制备了PVA/SA复合水凝胶。结果表明,复合水凝胶的形态和吸附性能与PVA/SA溶液质量比有关。当PVA(10wt%水溶液)/SA(3wt%水溶液)的比例为4∶1和3∶2时,样品表面表现出明显的网状结构。当水凝胶表面的吸附点过量时,其对Fe3+的吸附是无效的。水凝胶在前90 min内呈现出快速吸附模式。pH值的增加,有利于静电作用增强,使得水凝胶对Fe3+的吸附量增加。当PVA/SA溶液质量比为1∶4时,水凝胶对重金属离子表现出良好的吸附效果,对Fe3+的吸附量和吸附率分别达到83.11 mg/g和99.74%。准二级动力学模型表明其主要吸附机制是化学吸附。
猜你喜欢
空气动力学学报(2022年4期)2022-08-23
北京航空航天大学学报(2022年7期)2022-08-06
黑龙江大学自然科学学报(2022年1期)2022-03-29
上海金属(2021年6期)2021-12-02
昆明医科大学学报(2021年3期)2021-07-22
烟草科技(2021年6期)2021-06-24
陶瓷学报(2021年1期)2021-04-13
军事文摘(2020年20期)2020-11-16
陶瓷学报(2020年3期)2020-10-27
中学生数理化·八年级物理人教版(2020年12期)2020-01-01
