
甲醇气氛下低阶煤热解气中CO加氢制芳烃机理研究
2023-01-04 09:23:10王月伦安会会张雪纯马云欣詹贵贵刘豪杰曹景沛
燃料化学学报 2022年12期
王月伦 ,安会会 ,张雪纯 ,马云欣 ,詹贵贵 ,刘豪杰 ,曹景沛
(1.中国矿业大学 煤炭加工与高效洁净利用教育部重点实验室, 江苏 徐州 221116;2.中国矿业大学 江苏省碳资源精细化利用工程研究中心, 江苏 徐州 221116;3.徐州市建设工程检测中心有限公司, 江苏 徐州 221116)
轻质芳烃是重要的化工原料,广泛用于医药、染料、香精及高分子行业,尤以苯、甲苯、二甲苯(简称BTX)市场需求更高。传统的芳烃生产源于石油化工工艺,中国煤多油少的资源格局使煤炭的高效清洁利用日益受到重视[1,2]。热解是低阶煤分质利用的重点方向[3],热解提质是优化煤热解技术的有效途径[4]。
低阶煤热解过程释放大量的含氧芳香化合物、脂肪烃、热解气体等挥发分。这些组分可进一步转化为轻质芳烃,因而以低阶煤热解挥发分重整制备轻质芳烃受到研究者关注[5-10]。煤热解挥发物二次反应核心是C-O、C-C等键活化产生的自由基碎片与H·、CH3·等小分子自由基结合形成不同结构轻质产物。要实现活性物种有效匹配,热解气氛、催化剂是调控热解行为和产物组成的关键。目前,多采用催化热解以提高轻质芳烃选择性,催化剂有金属催化剂[5,7,9]、碳基催化剂[10]和分子筛催化剂[6-8]。其中,金属改性分子筛催化剂因其双活性中心协同使其成为重要的芳构化催化剂[11]。在催化过程中引入H2产生活性氢可稳定自由基碎片,提高轻质芳烃收率[12-14],但H2参与反应需高温高压,条件苛刻。外加供氢试剂通过氢转移是实现C-C键、C-O键活化转化的有效途径。靳立军等[15]采用甲烷芳构化与煤热解耦合,由于甲烷能产生大量 H·、CH3·、CH2·和 CH·自由基,与煤热解碎片耦合加氢显著提高了低碳芳烃产率。
前期Wang等[16]研究了甲醇气氛下低阶煤热解耦合挥发分原位重整制芳烃,发现甲醇气氛能显著提高轻质芳烃产率,归因于甲醇在热场生成了H·、CH3·等自由基。甲醇作为供氢试剂可实现C-C、C-O等键活化加氢,反应条件温和易控[5,6]。文献报道较多的是热解挥发分中酚类和醚类组分转化成单环芳烃[17,18]。由于低阶煤含氧官能团多,热解过程产生大量的CO和CO2,当体系存在活性氢,在催化剂作用下同样可实现CO或CO2加氢生成轻质芳烃。目前,关于甲醇气氛下低阶煤热解气中CO或CO2制备轻质芳烃的研究鲜有报道,而近年来,以合成气制备轻质芳烃引起了研究者广泛关注[19,20]。根据反应中间体不同,合成气制芳烃分为两种途径:一种为甲醇合成催化剂与分子筛耦合经中间产物甲醇制芳烃(SMA)[21];另一种为费托合成(FT)催化剂与分子筛耦合经过中间产物烯烃制芳烃(SOA)[22]。由于甲醇合成和甲醇芳构化反应条件不匹配,相比之下,费托合成反应和烯烃芳构化反应条件接近,更为适合合成气一步法转化为芳烃。与传统的合成气制芳烃不同的是本体系为甲醇气氛下低阶煤热解气中CO再转化。CO可与热解气中的H2转化制芳烃,同时甲醇经分子筛生成二甲醚,由C-C键偶联后生成低碳烯烃(),再经齐聚、环化和氢转移等也可转化为芳烃。其中,环化脱氢产生的活性氢易使烯烃加氢形成烷烃。如活性氢能及时转移,可避免烯烃加氢。研究发现[23],CO气氛可有效促进芳构化反应。H-ZSM-5分子筛上甲醇芳构化产生的H可迁移至金属活性中心,而H与吸附在金属活性中心的CO进一步反应,加速脱氢芳构化发生。因此,甲醇脱氢芳构化耦合CO加氢有望促进芳烃的生成。该过程可解决低阶煤热解组分提质过程轻质芳烃产率低的问题,可为低阶煤热解获取高附加值化学品提供技术支撑。目前,关于该过程制备轻质芳烃反应机理的理论计算鲜有报道。本工作基于Fe基催化剂对芳构化中间体烯烃有较高的选择性,将过渡金属Fe与HZSM-5分子筛耦合实现甲醇气氛下煤热解气中CO加氢制芳烃。采用密度泛函理论(DFT)探讨了CO在Fe/HZSM-5催化剂上加氢经由SOA过程生成芳烃可能的机理。
1 计算方法
铁基催化剂在FT反应过程中相态结构存在变化,目前,已检测到的活性相主要有金属铁、铁的氧化物以及碳化物。研究表明,CO加氢活性中心主要为碳化铁[23]。CO在Fe/HZSM-5催化剂上加氢制芳烃的反应可分为如下三步:在铁基催化剂上生成低碳烯烃C2H4,反应活性相主要为碳化铁(Fe5C2);低碳烯烃在HZSM-5中碳链增长生成;在HZSM-5中芳构化生成芳烃。
1.1 ZSM-5分子筛模型建立
由于小团簇模型具有原子个数少,对计算机配置要求低,计算量相对少等优点,且研究体系中参与反应的分子尺寸较小,在分子筛孔道中能与活性中心充分接触,因此,本研究选取了如图1所示由46个四面体单元组成的团簇模型ZSM-5,它包含了直孔道和正弦孔道以及孔道交叉处。ZSM-5分子筛的初始结构单元由Materials Studio数据库导入,通过截取得到46T团簇模型。截取过程中形成的悬断键由氢原子饱和,Si-H键长设置为1.47 Å,方向和初始模型中原来的Si-O键方向一致。ZSM-5分子筛的一个晶胞结构中含有12种不同的T位,铝原子由于周围环境的不同其稳定性不一样,T12被认为较为稳定。当Al原子取代T12位所在的Si原子时,形成带一个负电荷的四面体(),通过引入一个氢质子平衡此电荷,由此形成B酸中心位于孔道交叉处,作为反应的活性中心位置。
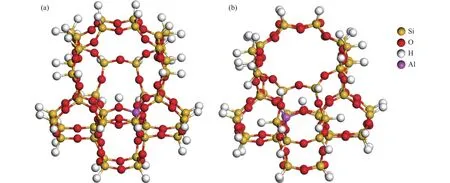
图1 46T团簇模型示意图Figure 1 Schematic diagram of 46T cluster model
1.2 Fe5C2 (510)模型建立
文献报道采用Wulffconstruction方法可获得Fe5C2微晶的平衡形貌及其暴露晶面的组成,表明Fe5C2(510)具有较低的表面能,且在暴露晶面中所占比例相对较高[24]。中间产物C2H4易于在Fe5C2(510)表面产生,因此,构建Fe5C2(510)模型。如图2所示,该模型包括40个Fe原子和16个C原子,其中底部四层C原子和两层Fe原子被固定,剩余的Fe原子和C原子位置弛豫。模型表面上真空层高度为 10 Å。计算时选用的模型大小为 P(2 × 1),k点值为(4 × 2 × 1),且考虑自旋极化效应。

图2 Fe5C2 (510)模型示意图Figure 2 Schematic diagram of Fe5C2 (510) model
所有计算均在Materials Studio软件包中CASTEP模块下完成。计算过程中,设置平面波基组截断动能为400 eV。第一布里渊区内的取样采用了Monkhorst-Pack的网格,设置smearing的宽度为0.2 eV。电子的交换关联能用广义梯度近似(GGA)的PBE泛函来进行描述。过渡态是基于反应物和产物结构采用LST/QST方法搜索。
2 结果与讨论
2.1 CO加氢生成C2H4过程
由CO加氢生成低碳烯烃C2H4包括CO解离、C1物种加氢、C1+ C1偶联,涉及四步反应[25]。其反应热、活化能、相对速率常数列于表1中。
CO解离:本研究计算采用CO直接解离机理。H和CO在Fe5C2(510)表面共吸附如图3所示,H吸附在3F-3位,吸附能为-75 kJ/mol,CO吸附在4F-1位,吸附能为-140 kJ/mol,H和CO在Fe5C2(510)表面吸附均为最稳定构型。随后CO和H开始扩散,CO通过TS1解离成C和O,图4显示的过渡态结构中,C-O键键长为1.96 Å。该步反应的活化能为124 kJ/mol,吸热反应其反应热为76 kJ/mol,其所需能垒最高,表明CO生成低碳烯烃决速步为C-O键的解离。

图3 CO和H吸附构型图Figure 3 CO and H adsorption configuration diagram
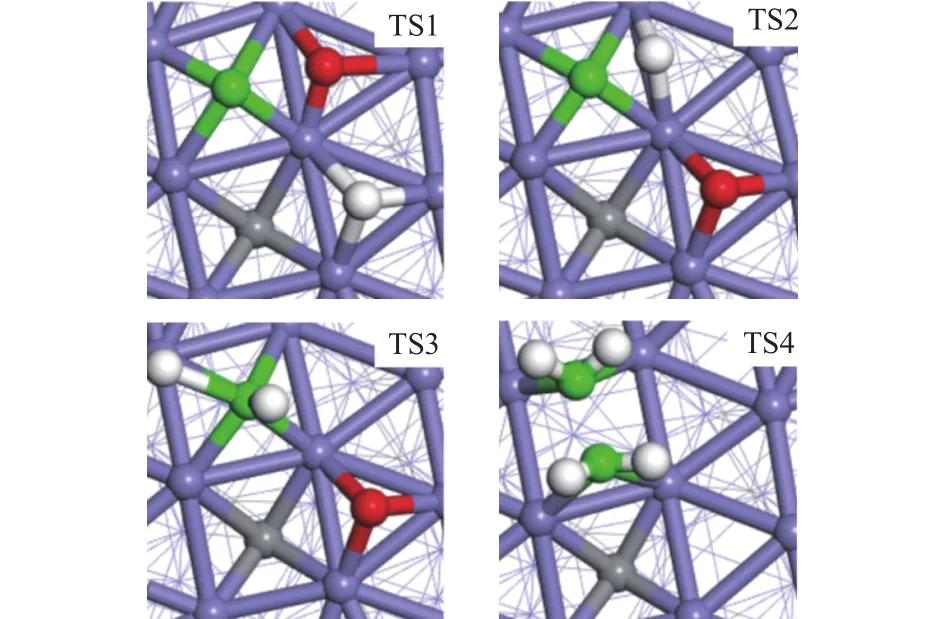
图4 生成中间体C2H4过程中的过渡态Figure 4 Transition states in the formation of C2H4
C1物种加氢:C1逐步加氢生成CH和CH2,这两步反应的活化能分别为12和82 kJ/mol,随着活性氢数目增加,由于CH2相对于CH更不稳定,加氢需要的过渡态能垒更高,反应速率也随之下降。在过渡态结构TS2和TS3中,即将形成的C-H键键长分别为1.43和1.92 Å。
C1+ C1偶联:目前为止,文献报道的链增长机制主要有三种:CO插入机理、碳化物机理和含氧中间体缩聚机理[26]。其中,碳化物机理已得到众多理论与实验验证,该机理认为,CH2是C1+ C1偶联链增长的单体。CH2偶联过程中过渡态结构TS4中C-C键键长为1.787 Å,此步反应活化能为112 kJ/mol,是吸热反应,反应热为 10 kJ/mol。
能量变化如图5所示,由图5可知,生成中间体C2H4过程中的过渡态以TS4势能最高,TS2势能最低,表明C-C键偶联实现碳增长最难,而活化后的C-O键再加氢形成CH中间体最容易。

图5 生成C2H4势能图Figure 5 Potential energies of the formation of C2H4
2.2 生成过程
甲醇气氛下芳构化过程长链烯烃形成是由低碳烯烃甲基化得到,同时多甲基苯又可经过甲基化消去产生低碳烯烃,为长链烯烃的生成提供了烯烃的来源,因而仍然遵循双循环机理[24]。图6为低碳烯烃在HZSM-5催化剂中碳链增长生成的可能路径。乙烯分子通过与甲醇不断发生甲基化(M1、M2、M3和 M4)和去质子化(D1、D2和 D3)形成较高碳链的碳正离子,其经历快速的分子内氢转移和甲基转移,最终生成己基碳正离子。

图6 生成路径示意图Figure 6 Path of the formation of
表2 生成C计算结果Table 2 Calculation results of forming (823 K,101 kPa)

表2 生成C计算结果Table 2 Calculation results of forming (823 K,101 kPa)
Reaction ΔH/(kJ·mol-1) ΔG≠/(kJ·mol-1) k/s-1 M1 30 135 4.37 × 104 D1 -42 53 7.25 × 109 M2 34 158 1.50 × 103 D2 -41 79 1.64 × 108 M3 51 96 1.35 × 107 D3 -22 61 2.31 × 109 M4 39 165 6.16 × 102

图7 生成路径势能图Figure 7 Potential energies of the formation of
甲基化:甲基化为乙烯和甲醇发生反应,脱去一分子水,生成碳正离子的过程[24]。四步甲基化的活化能分别为135、158、96与165 kJ/mol。甲基化过程速率常数值较低,表明反应速率较慢。
去质子化:碳正离子经历去质子化形成新的烯烃产物。此过程为放热反应,所需的活化能分别为53、79和61 kJ/mol,与甲基化过程相比,整体能垒降低,表明去质子化过程反应速率较快,容易发生。

图8 芳构化路径示意图Figure 8 Process of aromatization for
2-己基碳正离子与分子筛发生迅速地去质子化过程(D1)形成1-己烯,甲醇脱水后形成的甲基吸附于分子筛氧原子上形成烷氧基,1-己烯与生成的烷氧基之间发生氢转移(H1)得到烯烃碳正离子和甲烷,由于生成的烯烃碳正离子不稳定,发生快速的分子内氢转移得到异构化之后的烯烃碳正离子,为接下来的环化提供了优化构型,从而发生环化反应(C1)生成环己碳正离子。环己烷碳正离子经历同样的去质子化(D2)过程后,与分子筛内甲氧基发生第二步氢转移(H2)生成环己烯碳正离子。不稳定的环己烯碳正离子会继续发生去质子化(D3)和第三步氢转移(H3)生成环己二烯碳正离子,环己二烯碳正离子与分子筛发生最后一步去质子化(D4)生成了稳定产物苯。苯与甲醇不断提供的甲基可进一步甲基化进而生成甲苯和二甲苯等多甲基苯。
表3 芳构化计算结果Table 3 Calculation results of aromatization(823 K,101 kPa)

表3 芳构化计算结果Table 3 Calculation results of aromatization(823 K,101 kPa)
Reaction ΔH/(kJ·mol-1) ΔG≠/(kJ·mol-1) k/s-1 D1 -18 57 4.13 × 109 H1 60 122 3.10 × 105 C1 -109 78 1.92 × 108 D2 -55 48 1.54 × 1010 H2 -29 107 2.77 × 106 D3 -11 31 1.85 × 1011 H3 -27 94 1.85 × 107 D4 -81 15 1.91 × 1012
整个去质子化过程均为放热反应,所需的活化能在15-57 kJ/mol,其中,最小的速率常数为4.13 × 109s-1,表明在芳构化过程中,由不稳定的碳正离子去质子化形成己烯的反应相对容易,反应速率较快,而氢转移过程所需能垒较高,反应速率相对较慢。
甲醇脱水分解为甲基的过程为:甲醇分子首先在分子筛B酸活性位吸附活化,导致甲醇的C-O键断裂,然后形成甲基基团,并释放一分子水。其脱水过程过渡态如图10所示。此过程吸附的甲醇克服91 kJ/mol能垒脱水生成甲氧基,速率常数为2.87 × 107s-1,生成的水分子在下一步氢转移过程前脱附。

图10 甲醇脱水过程过渡态Figure 10 Transition state of methanol dehydration process
氢转移反应主要包括三步,第一步氢转移(H1)发生于2-甲氧基与1-己烯之间,甲氧基从1-己烯分子中夺取一个氢原子,生成甲烷,1-己烯生成新的碳正离子。此步反应的活化能为122 kJ/mol,图11所示的过渡态结构中,即将形成的CH4的C-H键键长为1.38 Å。第二步氢转移(H2)生成环己烯碳正离子,该过程的活化能为107 kJ/mol。生成环己二烯碳正离子的第三步氢转移(H3)的能垒为94 kJ/mol,速率常数为1.85 × 107s-1。在芳构化反应中,氢转移过程所需要的活化能最高,反应速率最慢,可以看出氢转移过程相对较难发生。在氢转移过渡态结构中,即将形成的CH4的C-H键键长在1.38-1.61 Å,反应物分子即将断裂的C-H键键长在1.35-1.38 Å,基本没有变化。环化反应(C1)是指直链烯烃碳正离子环化生成环烷烃碳正离子,此反应的过渡态能垒为78 kJ/mol,为放热过程其反应热为109 kJ/mol。

图11 芳构化氢转移过渡态Figure 11 Transition states of hydrogen transfer for aromatization
基于上述计算可知,相比传统的合成气制芳烃机理,H2在催化剂上吸附产生活性氢H,同时CO发生解离吸附进而与H生成CH2,CH2为链增长单体,通过不断的C-C键偶联实现链增长,随后通过齐聚、环化和氢转移形成芳烃。本研究为甲醇芳构化过程中脱氢,产生的活性氢与CO反应生成CH2,进而实现C-C键偶联形成低碳烯烃,随后与甲醇在分子筛上甲基化实现碳链增长,生成高碳离子,再环化脱氢芳构化生成芳烃。因而在此过程中甲醇不仅提供活性氢,还提供甲基。两者耦合促进了轻质芳烃生成。
3 结 论
本研究采用DFT方法研究了甲醇气氛下低阶煤热解气CO于Fe/HZSM-5催化剂上加氢经烯烃中间体制芳烃机理。结果表明,甲醇气氛下CO可于Fe5C2(510)表面加氢生成低碳烯烃,其CO解离为该过程的速控步。通过多次甲基化和去质子化过程C-C键偶联实现链增长,其中,甲基化需较高的活化能。芳构化过程经过氢转移、去质子化及环化(C)过程,其中氢转移最难,反应速率常数最低。整个CO加氢制芳烃过程以甲基化所需能垒最高,成为该反应的决速步。总之,甲醇通过供氢或甲基为低阶煤热解气中CO定向转化制轻质芳烃提供了一条新途径。
猜你喜欢
大学化学(2023年11期)2024-01-23 12:55:02
——碳正离子的产生及稳定性比较
高中数理化(2022年20期)2022-11-17 02:42:28
数理化解题研究·高中版(2022年10期)2022-05-30 23:30:12
天津化工(2020年5期)2020-10-15 07:38:54
当代化工(2019年10期)2019-12-02 01:25:01
西安文理学院学报(自然科学版)(2016年4期)2016-12-19 08:18:59
China International Studies(2016年3期)2016-07-14 03:00:06
化工进展(2015年3期)2015-11-11 09:09:44
质谱学报(2015年5期)2015-03-01 03:18:25
河南科技(2014年3期)2014-02-27 14:05:47
