
多种碳正离子的结构和稳定性问题的探讨
2024-01-23 12:55:02王文峰袁耀锋
大学化学 2023年11期
王文峰,袁耀锋
福州大学化学学院,福州 350116
1 引言
碳正离子是《有机化学》上册教学中最重要的中间体,其结构和稳定性一直是物理有机化学家最感兴趣的研究内容[1,2]。由于课时限制,多数高校在碳正离子的教学中只能介绍一些已有定论的简单知识,不能满足今后有志于从事有机化学研究工作的学生的需求。本文计算了多种碳正离子的结构和稳定性问题,旨在使大学生对这个中间体有一个更深入的了解。
2 计算方法
本文采用Gaussian 03[3]程序和密度泛函理论(DFT)中的B3LYP方法计算所有化合物的分子结构,对所有原子采用6-311+G**基组并用全优化方法计算其稳定结构。
3 碳正离子结构和稳定性
3.1 碳正离子的超共价结构
许多权威的有机化学教科书[4,5]都介绍碳正离子为sp2杂化,碳形成三个共平面的共价键。绝大部分碳正离子的结构的确如此,但是有例外情况。物理有机化学家的研究证实[6]51-52(中括号外为页码,后同),乙基碳正离子的5个H原子无论在溶液中还是在气相中都以很快的速率进行重排。这种现象表明碳正离子很可能具有如Scheme 1所示的动态结构,但是在动态结构中,桥形结构和开环结构哪一个具有更低的能量,物理有机化学家不能确定。
本文采用DFT方法对乙基碳正离子的结构和稳定性进行了计算。DFT方法优化乙基碳正离子的结构时,依据初始结构的不同,可以得到两个不同的乙基碳正离子结构。第一种初始结构为:甲基中有一个C—H与碳正离子所在平面共平面,如图1A所示,C1—H3键与碳正离子所在平面共平面,同时这个平面均匀分开C1—H4和C1—H5两个共价键。在这种结构中,没有一个H处于可迁移的位置,所以本文称之为不迁移初始结构。第二种初始结构为:甲基中有一个C—H键与碳正离子的未杂化p轨道共平面,如图1B的C1—H3键,这种结构就是Scheme 1中的开环结构。这种构型中C1—H3键可以和碳正离子发生超共轭效应,且H3原子可以迁移到邻近的碳正离子上,本文称之为可迁移初始构型。这两种初始构型优化后的结构分别显示于图1C和图1D。
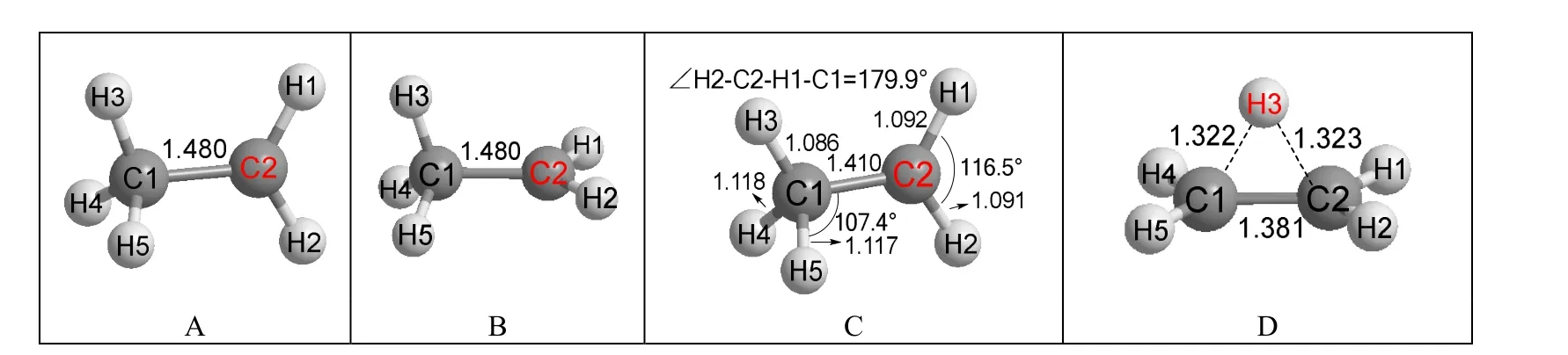
图1 乙基碳正离子的初始构型和优化后结构
从不迁移初始构型的优化后结构图1C来看,优化后的确没有H原子发生迁移,碳正离子也的确是平面型的,因为二面角H2—C2—H1—C1为179.9°。甲基中的三个C—H键长有差别,C1—H3键因为与碳正离子共平面,与碳正离子完全没有超共轭效应,所以键长只有0.1086 nm。C1—H4和C1—H5不与碳正离子共平面,有部分超共轭效应,键长被部分拉长,分别为0.1118和0.1117 nm。
可迁移初始构型优化后H3原子的确发生了迁移,得到了桥形结构图1D。在图1D中,C—C键长只有0.1381 nm,两个碳原子和四个氢原子非常接近共平面,同时H3原子与两个碳原子的键长都在0.1322 nm左右,比正常C—H键长得多。仿佛一个H+浮在一个平面乙烯分子上方。这种结构中H3原子同时形成了两个共价键,与Scheme 1中的桥型结构相同,这种H原子被称为超共价原子[6]51-52,本文将这种含有超共价原子的结构称为超共价结构(Super covalent structure)。以超共价结构图1D的能量为零,则不迁移初始构型优化后结构图1C的能量为15.7 kJ·mol-1。不迁移结构和可迁移结构都属于开环结构,可迁移结构优化后得到的是桥型结构(即超共价结构),而不迁移结构优化后能量高于桥型结构,这些结果说明Scheme 1中桥型结构比开环结构更稳定。
3.2 丙基碳正离子的结构
是不是所有碳正离子的最稳定结构都是超共价结构呢?为回答这个问题,本文选择对丙基碳正离子进行计算。采用类似图1的方式,给出两种初始构型分别进行优化计算,结果如图2所示。
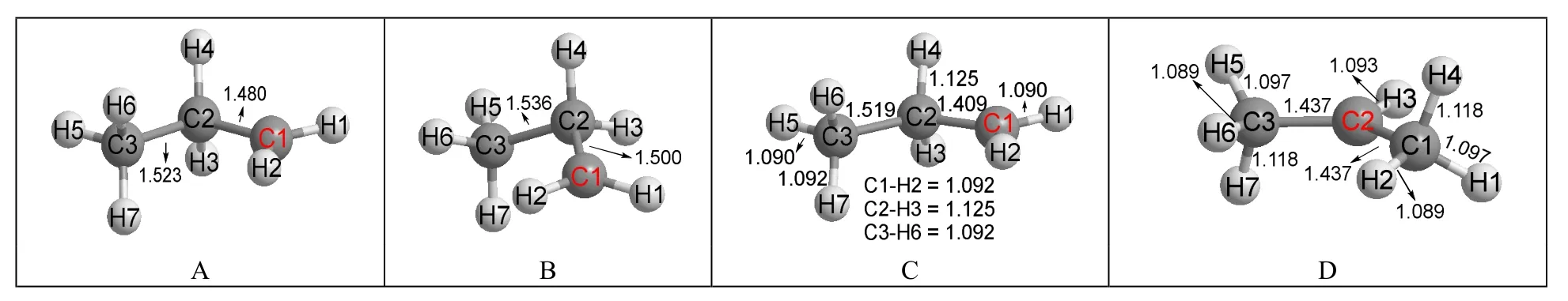
图2 丙基碳正离子的初始构型和优化后结构
在图2中,两个初始构型(图2A和图2B)显示的都是正丙基碳正离子的结构,即。与图1的计算结果类似,不迁移初始构型优化后没有发生H原子的迁移,得到的仍然是正丙基碳正离子的结构(图2C)。图2C结构中C2—H4和C2—H3的键长均为0.1125 nm,比其余C—H键更长,表明这两个C—H键与相邻的碳正离子存在超共轭效应。另外,图2C结构中C2—C1的键长(0.1409 nm)比C2—C3的键长(0.1519 nm)短得多,原因是超共轭效应的存在使得C2—C1键具有双键性质,如Scheme 2所示:
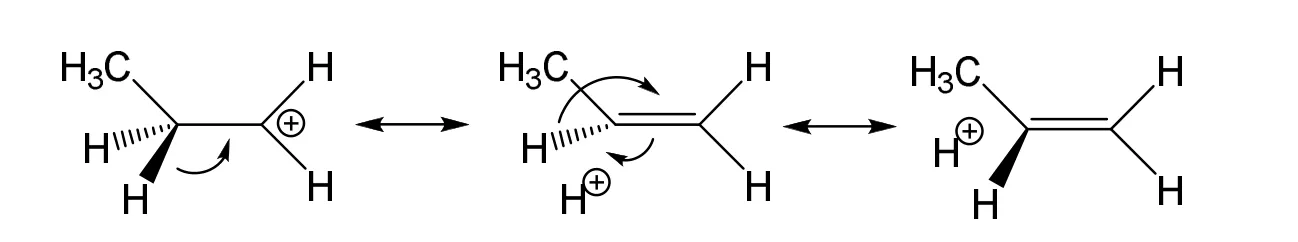
Scheme 2 丙基碳正离子通过无键共振形成超共轭效应的结构表示图
在可迁移初始构型优化后的结构图2D中,H4原子的确发生了迁移,得到的是异丙基碳正离子的结构,即CH3CH+CH3。以图2D结构的能量为参照(即设为0.0 kJ·mol-1),则图2C结构的相对能量是61.6 kJ·mol-1。这个结果表明仲碳正离子比伯碳正离子稳定得多,该结论与大学教科书结论一致。
乙基碳正离子之所以采用超共价结构,一个重要原因是它的两个经典结构是等价的。但丙基碳正离子的两个经典结构分别是正丙基碳正离子和异丙基碳正离子(以H原子为迁移原子),能量相差很大,完全不等价,所以其最稳定结构是异丙基碳正离子而不是超共价结构。异丙基碳正离子具有对称结构(见图2D),左右两个甲基是等价的,所以其结构中显示了等价的超共轭效应:C3—H7键和C1—H4键都和碳正离子未杂化的p轨道共平面,它们与碳正离子的超共轭效应最强,键长都是0.1118 nm,在所有C—H键中最长;C3—H6键和C1—H2键由于与碳正离子所在平面几乎共平面,这两个C—H键几乎没有与碳正离子发生超共轭作用,它们的键长0.1089 nm是所有C—H键中最短的。C3—H5和C1—H1则由于与碳正离子存在程度很小的超共轭作用,所以它们的键长值0.1097 nm处于中间。此外,C2正离子和C1及C3两原子的键长都是0.1437 nm,而叔丁基碳正离子[(CH3)3C+]的晶体结构中碳碳键长是0.1442 nm[6]50,两者十分接近,表明本文的计算结果可靠。
3.3 仲丁基碳正离子的结构
丙基碳正离子因为具有不等价的经典结构而不采取超共价结构,那么是不是具有等价经典结构的碳正离子都会像乙基碳正离子那样以超共价结构作为最稳定结构呢?为了回答这个问题,本文选择对仲丁基碳正离子进行结构优化。因为仲丁基碳正离子的两个经典结构是等价的,如Scheme 3所示。本文以可迁移构型为初始构型,考察H原子是否会迁移形成超共价结构。计算结果列在图3。
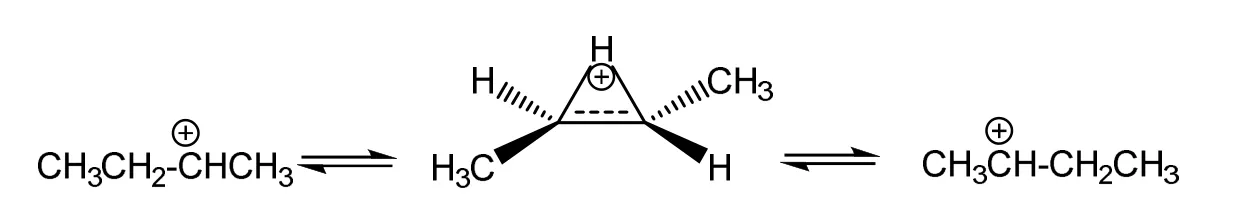
Scheme 3 仲丁基碳正离子的超共价结构和经典结构
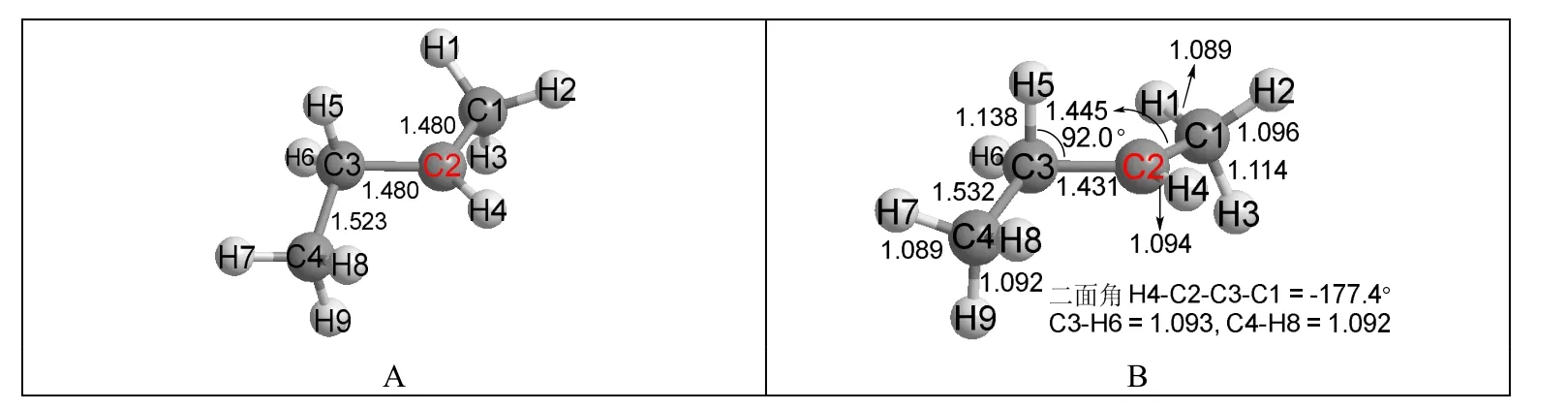
图3 仲丁基碳正离子的结构
在图3A中,H3和H5原子都处在可迁移的位置。但是经过优化后,这两个原子都没有发生迁移。而且仲丁基碳正离子与异丙基碳正离子不同,它不是对称结构,C2正离子与相邻的CH3和CH2上的C—H键的超共轭效应不等价,此时如何选择?从图3B的结构中能发现如下几点:(1) C2—C3的键长0.1431 nm短于C2—C1的键长0.1445 nm,表明C2—C3比C2—C1具有更多的双键成分。原因是C2—C1形成的双键是末端双键,超共轭效应弱,而C2—C3双键是链内双键,超共轭效应强;(2) C3—H5的键长0.1138 nm比C1—H3的键长0.1114 nm长,表明C3—H5与碳正离子的超共轭效应比C1—H3强;(3) H5—C3—C2的键角只有92.0°,远远偏离了sp3杂化碳原子的正常键角109.5°,是因为接近90°的键角能和碳正离子的空p轨道更好地发生重叠;而C2—C1—H3的键角是103.0° (图3中未列出),进一步表明C3—H5的超共轭效应强于C1—H3的超共轭效应。这些结果指向一个结论:由于形成双键是发生超共轭作用的附带结果(如Scheme 2所示),所以碳正离子总是倾向于与能形成更稳定双键的碳原子上的C—H键发生超共轭作用,而且这种超共轭作用能较大程度改变C—H的键长和键角。
3.4 仲碳正离子到叔碳正离子的重排
3.2部分的计算表明伯碳正离子优化后会得到仲碳正离子,那么仲碳正离子优化后是否会得到叔碳正离子?本文以可迁移构型为初始构型,对3-甲基-2-丁基碳正离子进行了优化,结果见图4。
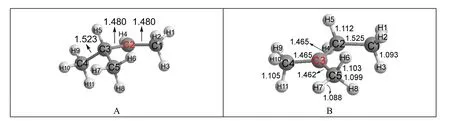
图4 3-甲基-2-丁基碳正离子的结构
由图4可见,优化后H5原子确实迁移到C2原子上,C3成为一个叔碳正离子。这表明仲碳正离子在优化时也会重排成更稳定的叔碳正离子。结合3.2部分的计算,可以得出一个推论:如果一个碳正离子能够重排成更稳定的碳正离子,则这种重排反应非常容易发生。
3.5 以碳为超共价原子的碳正离子
在前述的所有讨论中,本文都以H原子作为迁移基团。如果以CH3作为迁移基团,能不能获得以碳为超共价原子的碳正离子呢?如果能,这样的碳正离子是否能比以H原子为超共价原子的碳正离子稳定?为了回答这个问题,本文选择将CH3作为迁移基团来优化正丙基碳正离子CH3CH2CH2+的结构。结果列于图5。
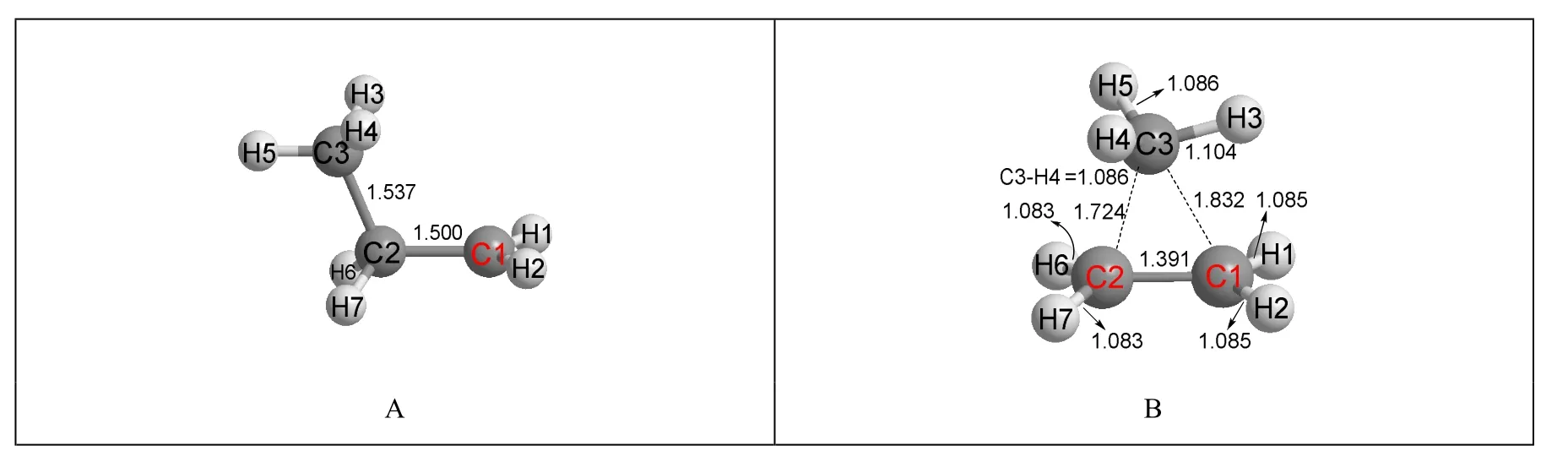
图5 以甲基为迁移基团的丙基碳正离子的结构
在优化后的结构图5B中,甲基确实发生了迁移,形成了一个以碳原子为超共价原子的超共价结构。但与乙基超共价结构图1D有区别的是,下方的两个碳原子(C1和C2)与四个氢原子(H1、H2、H6和H7)不共平面,且C3原子不处于C1和C2两个原子的中间位置,即C3—C2与C3—C1的键长不相等。但整个分子有对称面,C1、C2、C3与H3这四个原子共平面,且将分子中剩余的6个氢原子平分在平面两端,所以该平面即分子的对称面。与H作为迁移基团的优化后结构图2D的能量相比,若图2D的能量为0.0,则图5B的能量为44.15 kJ·mol-1,远远不如图2D稳定。这说明以碳为超共价原子的超共价结构不是碳正离子的稳定结构,同时也表明以氢原子作为可迁移基团来进行碳正离子结构优化的方法是正确的。
3.6 烯丙型碳正离子的稳定性问题
在基础有机化学教科书中,烯丙型碳正离子和叔碳正离子都属于非常稳定的碳正离子,但没有介绍哪一种碳正离子更稳定。在高等有机化学教科书中,用氢负离子的亲和性(定义为氢负离子与碳正离子结合为相应烷烃所放出的能量,HIA)来衡量气态碳正离子的稳定性[6]85-86,表1列出一些气态碳正离子的HIA值。
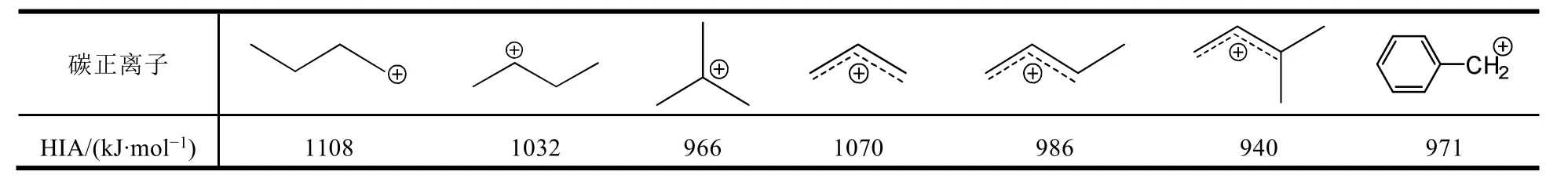
表1 丁基碳正离子与烯丙基型碳正离子和苄基碳正离子的HIA值
HIA值越小,碳正离子越稳定。表1列出的三个烯丙基碳正离子都是伯烯丙基碳正离子。没有取代基的烯丙基碳正离子的HIA是1070 kJ·mol-1,稳定性还不如仲碳正离子。有一个和两个取代基的伯烯丙基碳正离子的HIA分别是986和940 kJ·mol-1,已经分别比仲碳正离子(1032 kJ·mol-1)和叔碳正离子(966 kJ·mol-1)稳定。这表明烯丙型碳正离子随取代基的不同,稳定性有很大差别。
本文优化计算了如图6所示的五种同分异构体,依据碳正离子和双键位置的不同,它们分别属于仲碳正离子、叔碳正离子、一取代伯烯丙型碳正离子、仲烯丙型碳正离子和叔烯丙型碳正离子。
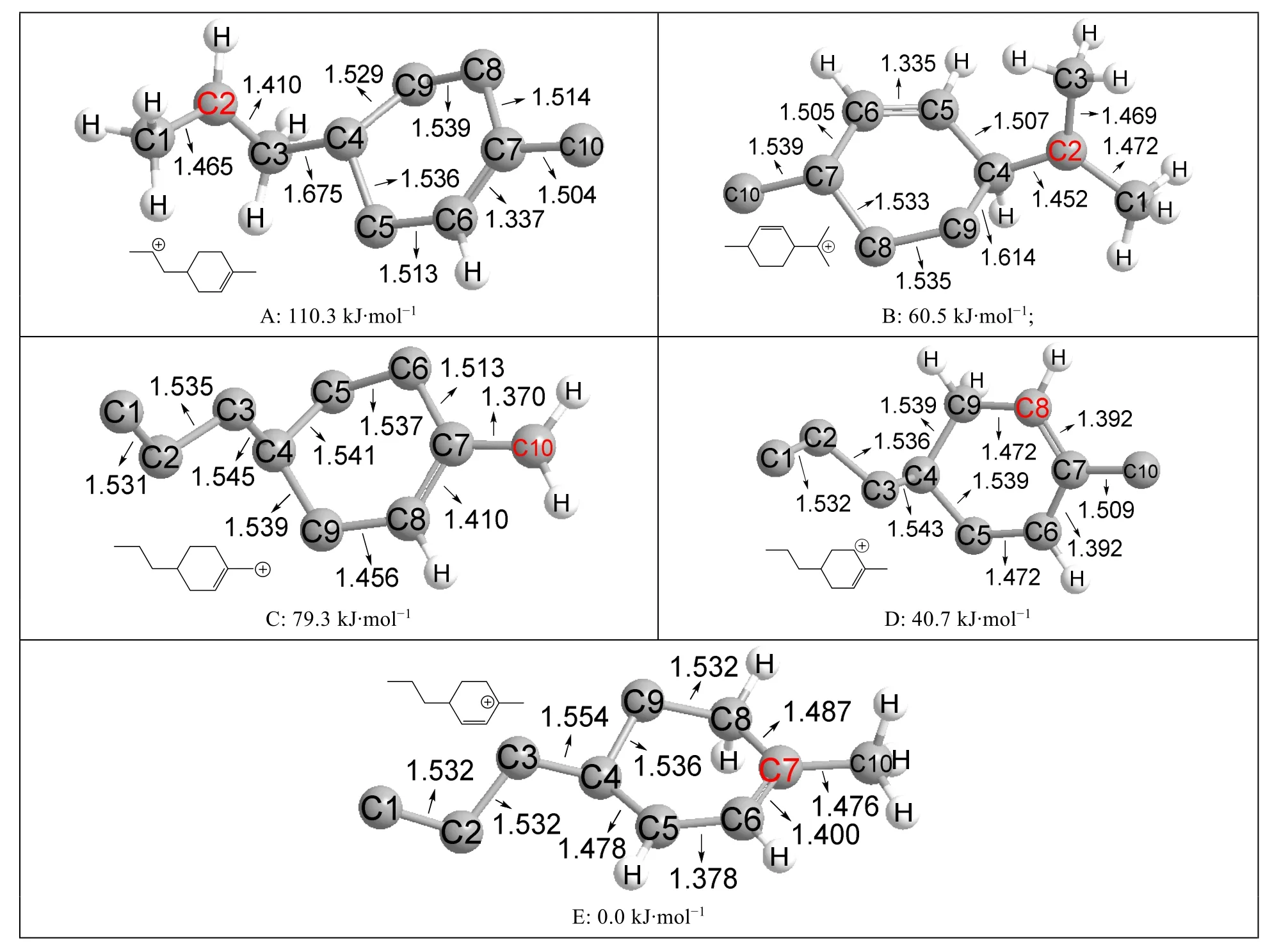
图6 五种碳正离子同分异构体优化后的结构图及其相对能量
以最稳定的叔烯丙型碳正离子为参照,其能量设为0.0 kJ·mol-1。从图6中可以看出,叔碳正离子的稳定性比仲碳正离子稳定得多,能量差约50 kJ·mol-1;一取代伯烯丙基碳正离子比仲碳正离子稳定,但不如叔碳正离子稳定,与表1的计算结果相同。仲烯丙型碳正离子已经比叔碳正离子稳定,叔烯丙型碳正离子则比仲烯丙型碳正离子更稳定,两者能量差约40 kJ·mol-1。
在图6D的结构中,C7—C6和C7—C8的键长都是0.1392 nm,表明碳正离子和双键已经完全离域。类似地,图6C和图6E的结构中碳正离子和双键也存在明显离域。所以烯丙型碳正离子的稳定性可以归因于共轭效应。
3.7 一些特别稳定的碳正离子
具有芳香性或者α-位含有羟基、氨基的碳正离子是特别稳定的碳正离子,如片呐醇重排的驱动力是叔碳正离子重排为α-羟基碳正离子,表明α-羟基碳正离子比叔碳正离子还稳定。为了定量衡量这类碳正离子的稳定性,本文计算了如下7个碳正离子,它们分子中的共价键类型和数量都相同,所以能量差别主要来自于碳正离子的稳定性差别,结果见图7。
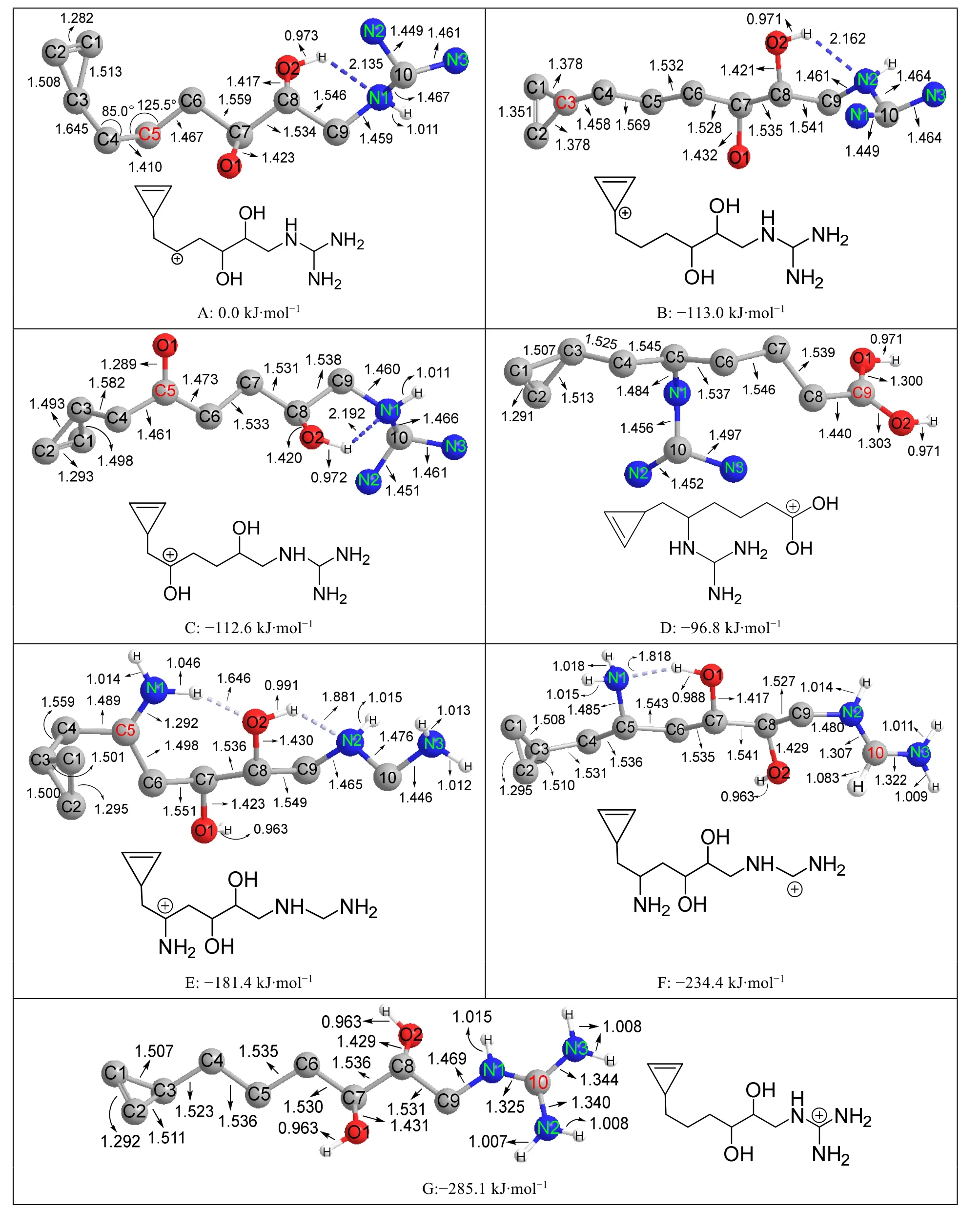
图7 互为同分异构体的7个碳正离子的结构和能量
从图7可以得到如下结果:
1) 以仲碳正离子的能量为参照(设为0.0 kJ·mol-1),可以看出其他6个碳正离子都比仲碳正离子稳定得多,它们与仲碳正离子的能量差值(约100-285 kJ·mol-1)都明显超过叔碳正离子与仲碳正离子的能量差值(约50 kJ·mol-1),说明这些碳正离子都比叔碳正离子稳定得多。
2) 芳香正离子(本文为环丙基碳正离子)与单羟基碳正离子的稳定性相当。氨基碳正离子比单羟基碳正离子稳定得多,表明氨基对碳正离子的稳定作用明显超过羟基。每增加一个氨基,化合物的能量降低约50 kJ·mol-1,所以脒盐和胍盐碳正离子都非常稳定,与实验上发现胍和脒都是强碱的事实吻合。但是双羟碳正离子(图7D)反而不如单羟碳正离子(图7C)稳定,原因可能是单羟碳正离子中有一个氢键作用(N1…HO2)而双羟碳正离子没有(图7D中没有氢键作用)。为了排除氢键数目不等造成的干扰,本文又计算了互为同分异构体且都没有氢键作用的两个碳正离子的能量,结果列于图8。图8中双羟碳正离子(图8B)比单羟碳正离子(图8A)稳定约7.4 kJ·mol-1,这个计算结果与羧酸的碱性(质子化乙酸pKa= -6.1)强于醛酮(质子化丙酮pKa= -7.3)的实验事实吻合。根据上述pKa差值估算的(ΔG= -2.303RTlgKa1/Ka2,按照Ka1/Ka2= 101.2、T= 300 K估算)质子化乙酸(具有双羟碳正离子结构)和质子化丙酮(具有单羟碳正离子结构)稳定性差别约:2.303 × 2.493 × 1.2 = 6.9 kJ·mol-1,这个值与本文计算值(7.4 kJ·mol-1)非常接近,表明B3LYP方法在计算化合物能量方面比较可靠。由于双羟基碳正离子只比单羟基正离子稳定7.4 kJ·mol-1,所以一个氢键的差别(氢键键能约20-25 kJ·mol-1)就足以使7C反而比7D稳定15.8 kJ·mol-1。
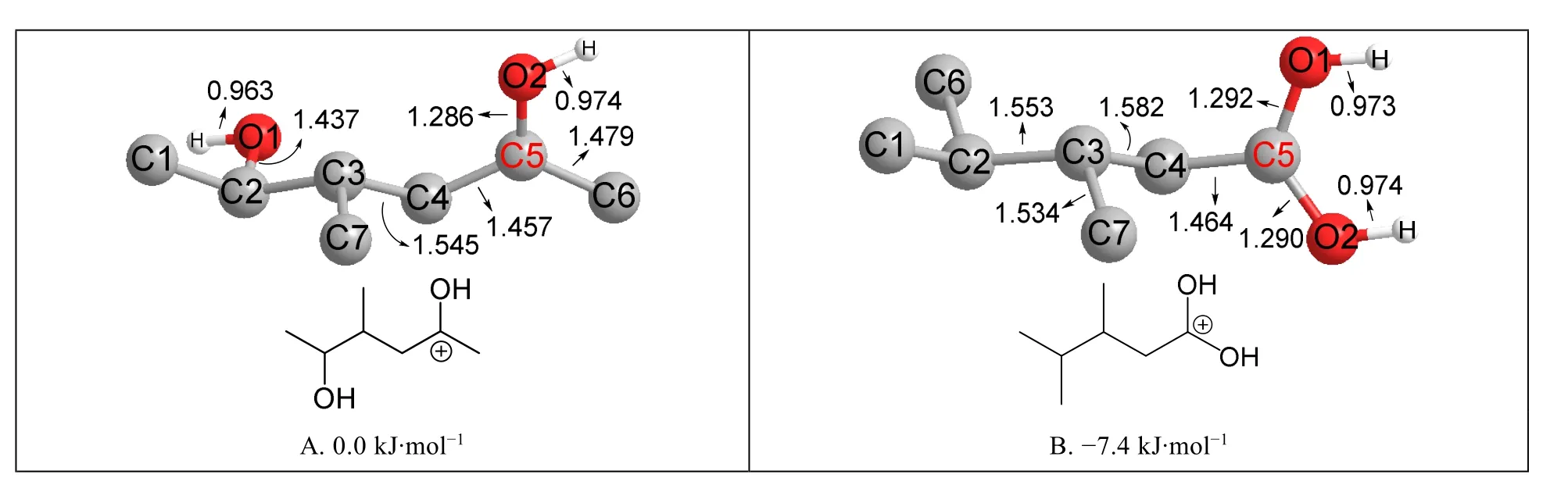
图8 羟基碳正离子的结构和相对能量
3) 当NH2与OH位置靠近时,既可以形成O—H…N氢键,也能形成N—H…O氢键,从图7A、7B、7C、7F看,形成的都是前者而不是后者,表明前者是比后者更强的氢键。从图7F和图7G来看,脒盐或胍盐中的氨基因为其孤对电子与碳正离子有强烈共轭效应,N原子的电子密度大大降低,已经不能再与邻近的OH形成氢键,表明氢键的形成对受体的电子密度有较高要求。
4) OH和NH2在形成氢键时,O—H和N—H键长都会拉长,前者大约从0.0963 nm拉长到0.0988 nm,后者大约从0.1012 nm拉长到0.1046 nm。但是与碳正离子形成共轭效应后(即与碳正离子紧相邻),O—H和N—H的键长变化方向不一样。普通OH中O—H键长约0.0963 nm,与碳正离子共轭后拉长到0.0971 nm,如图7D所示。普通NH2中N—H键长约0.1012 nm,与碳正离子共轭后缩短到0.1008 nm,如图7G所示。导致这种现象的原因可能是OH与碳正离子共轭后呈现质子化酮的结构,质子化酮酸性很强,容易电离出H+,故O—H变长;但是NH2与碳正离子共轭后呈现质子化亚胺的结构,质子化亚胺酸性不强,不易电离出H+,NH2只好通过调整杂化状态来加强共轭效应,从普通NH2的sp3杂化调整为sp2杂化,以便孤对电子所在的未杂化p轨道可以与碳正离子的未杂化空p轨道能更好重叠。由于sp2轨道的s成分比sp3高,更加收缩,故N—H键长较短,如同烯烃的C—H键长(通常约0.1085 nm)比烷烃的C—H键长(通常约0.1095 nm)略短一样,如Scheme 4所示。对于双羟碳正离子,第一个羟基氢原子的电离对第二个羟基氢原子的电离有抑制作用,所以第二个羟基对相邻碳正离子的稳定作用不如第一个羟基大,导致双羟碳正离子仅比单羟碳正离子稳定7.4 kJ·mol-1,不像脒盐正离子与氨基碳正离子的稳定性差别能达到53.0 kJ·mol-1。
5) 图7A中C3—C4的键长是0.1645 nm,图7B中C3—C4的键长只有0.1458 nm,分别是图7所有化合物中最长和最短的C—C单键(普通的C—C单键键长为0.1540 nm)。这种结构现象可以通过碳正离子的共振得到解释,如Scheme 5所示。其中左图仲碳正离子的共振结构还能解释图7A中∠C3—C4—C5只有85.0°的反常现象。
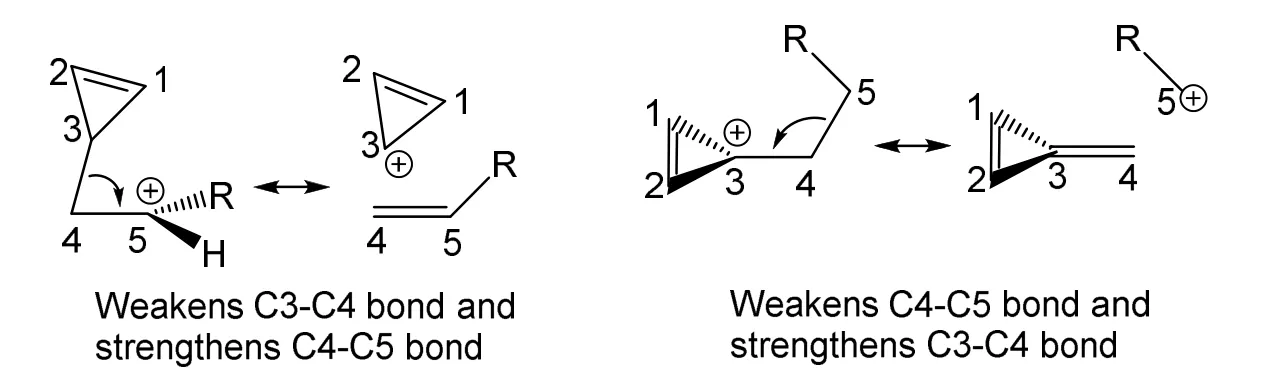
Scheme 5 图7A (左)和图7B (右)所示碳正离子的共振结构图
4 结语
伯碳正离子比仲碳正离子不稳定得多,能量差值约60 kJ·mol-1;仲碳正离子又比叔碳正离子不稳定得多,能量差值约50 kJ·mol-1。普通的伯碳正离子在优化时总是重排为仲碳正离子,但乙基碳正离子是例外,因为它无法重排为仲碳正离子,所以采用超共价结构来避免形成伯碳正离子。仲碳正离子相对稳定,当它们不能重排为叔碳正离子时,它们也不形成超共价结构,而是形成强的超共轭效应。烯丙基型碳正离子都比较稳定,优化时不发生重排。其中仲和叔烯丙型碳正离子比叔碳正离子稳定。α-位有羟基或者氨基的碳正离子特别稳定,α-羟基碳正离子的稳定性与具有芳香性的环丙烯碳正离子相当,比叔碳正离子稳定约50 kJ·mol-1。α-氨基碳正离子比α-羟基碳正离子更稳定,能量差值约70 kJ·mol-1。在α-位每增加一个氨基,碳正离子能量约降低50 kJ·mol-1。但是α-位增加一个羟基对碳正离子能量的降低作用很有限,只有约7.4 kJ·mol-1。氨基和羟基的这个差别可能源自它们对碳正离子的不同的稳定作用机制。
猜你喜欢
中国感染控制杂志(2022年10期)2022-11-24 02:18:46
——碳正离子的产生及稳定性比较
高中数理化(2022年20期)2022-11-17 02:42:28
数理化解题研究·高中版(2022年10期)2022-05-30 23:30:12
潍坊学院学报(2021年2期)2021-07-22 07:59:12
高等学校化学学报(2021年7期)2021-07-11 16:25:48
天津化工(2020年5期)2020-10-15 07:38:54
东华大学学报(自然科学版)(2018年1期)2018-06-29 03:34:54
中国医药指南(2017年3期)2017-11-13 02:55:35
卫生职业教育(2014年11期)2014-05-27 08:38:08
安徽农业科学(2014年27期)2014-04-29 18:47:11
