
浅议分子编辑
——原子尺度上的积木游戏
2024-01-23 12:55:14周钰翔谭杨昊宇叶子瑞王铭邦卞江
大学化学 2023年11期
周钰翔,谭杨昊宇,叶子瑞,王铭邦,卞江
北京大学化学与分子工程学院,北京 100871
分子编辑技术,是直接在分子原有的结构基础上对其进行的原子尺度上的修饰(图1),能极大程度地简化合成路线与逆合成分析的难度。自分子编辑技术出现以来,有机合成化学家们主要围绕在两个方面不断优化试剂和思路:外围编辑(peripheral editing)和骨架编辑(skeleton editing)。
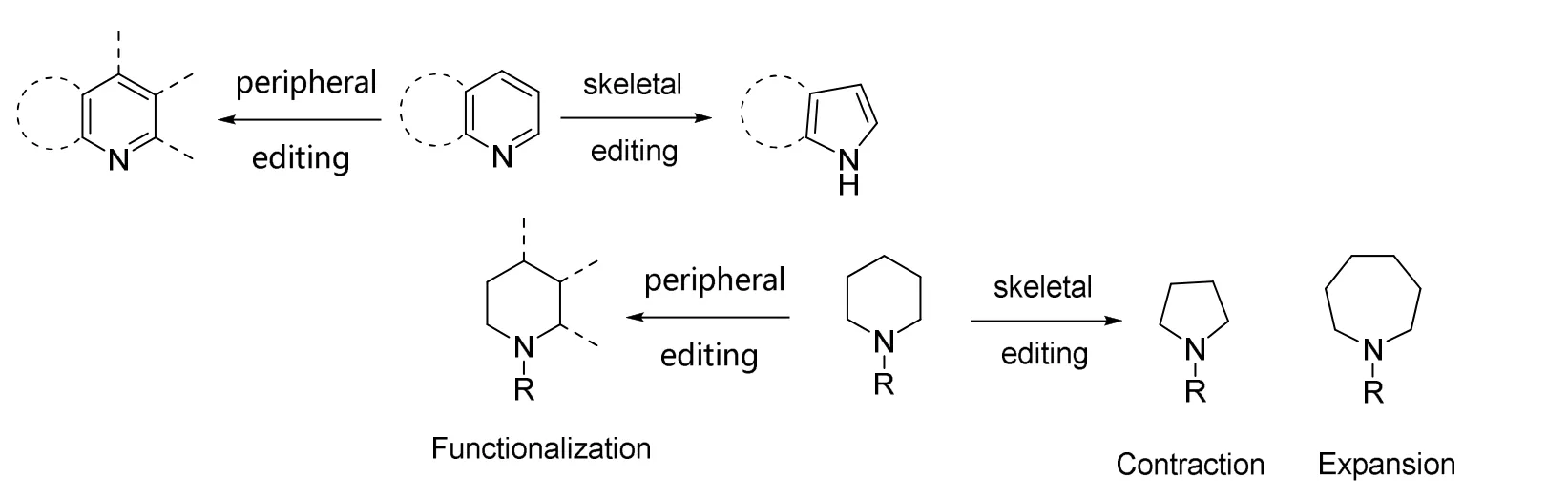
图1 分子编辑反应的示意图
外围编辑主要关注在催化剂或其他条件下对惰性的C—H进行的活化,从而导入其他目的基团,实现从无到有的突破,在温和的条件下将目标分子官能化。这样的策略克服了C—H在剧烈条件下才容易发生化学反应的问题,在通过电子效应和空间效应控制反应位点的同时还可以利用不对称配体控制产物的手性。到目前为止,外围编辑技术仍主要围绕C—H活化实现直接官能化[1]。此外,也有工作报道了将C—H活化构建为C—B实现间接官能化,从而实现间接的分子外围编辑[2]。
近年来,骨架编辑领域的新进展吸引了许多有机化学家的关注。尽管C—H官能化在合成上是一种相对有效的策略,但其并没有充分利用分子骨架的巨大潜力。骨架编辑以重排和骨架跃迁为中心,在光照、特殊试剂的作用下,通过单个原子的删除和插入来进行分子骨架的调整,从而实现分子的快速合成,这样可以减少步骤,节省原料,降低成本。已经得到发展的重排反应如Baeyer-Villiger氧化、Hoffman重排,尽管也是“单原子操作”,但由于反应的局限性和反应条件可能导致的兼容性问题,其在有机合成中的应用范围也不尽相同。如何开发一种普适的骨架编辑策略也许会成为未来的主要研究方向。本文将主要围绕外围编辑和骨架编辑两方面展开讨论。
1 外围编辑(peripheral editing)
1.1 外围编辑的主要策略
外围编辑对分子骨架外围的C—H进行修饰,得到官能化的产物。比如利用金属钯可以直接对目标的C—H片段进行处理,插入芳基或硼化等。这样可以将惰性的C—H键转化为有反应活性的其他基团,为后续反应激发了无限可能。
1.1.1 直接官能化
通常的C—H活化是在定向基团的引导下,结合C—H的弱配位作用,将钯引导至合适的空间位置(图2),与目标位置的C—H发生金属的插入反应得到中间体,然后与其他反应物反应得到产物(图3)。而余金权教授等在2014年报道了使用钯催化剂,在底物中存在氰基(导向基团)时发生远程C—H活化的反应[3]。
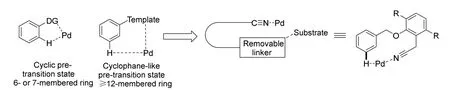
图2 远程C—H官能化的示意图
其前过渡状态处于12元环的状态,远大于正常情况下的6-7元环,其好处是不必在反应位点附近再引入导向基,而原本作为导向基的氰基还可以在后期的反应中进一步转化为羧基、羰基,或氨基以及相关的其他官能团。
2016年,余金权教授等再次在Science上报道了通过手性配体对钯进行配位,对羰基β位进行芳基化的反应[4]。通过不断改进配体的结构,得到了同时具有较高的对映选择性与良好产率的催化剂(图4)。产物中同样的β-芳基化结构一般需要通过金属铑催化的不对称共轭加成得到[5],而这一反应为同样的结构提供了新的逆合成思路。
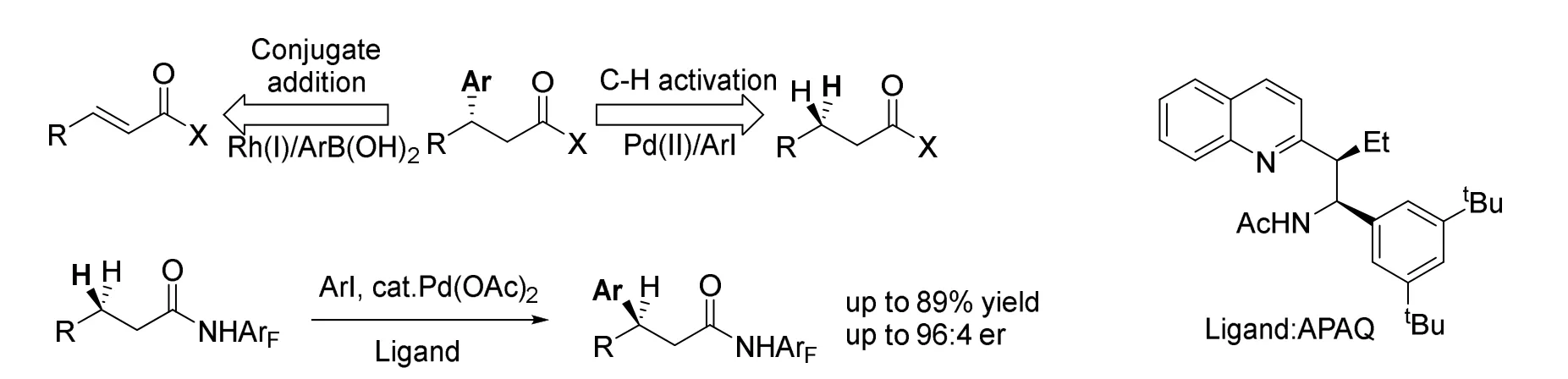
图4 Pd引导的不对称C—H活化
上述的系列反应主要通过C—H的弱配位作用与杂原子的配位进行导向,这要求事先在特定位置引入导向基团。2022年,余金权等再次在Nature上报道了通过控制配体大小,利用空间效应对C—H进行选择性活化的反应[6]。此反应最独出心裁的一点,就是绕开了传统的“要对一个特定位点进行活化就要先引入导向基”的观念,改变配体的结构从而使金属能够与不同的碳氢键进行反应(图5),某种程度上来说,延续了远程碳氢活化的思想。
图5 空间效应引导的C—H活化
1.1.2 间接官能化
同样是C—H活化,直接官能化通常只进行烯基化、芳基化等反应,难以进行羟基、醛基等官能团的引入,我们推测原因是这类官能团具有高配位活性的氧原子可以与中心金属进行配位,从而阻碍目标反应的发生。为了在想要的位点引入这类官能团,一种已知的策略是先将C—H转化为C—B键,再通过Suzuki偶联或硼氢化-氧化等一系列反应进行进一步转化,得到更为广泛的产物。这也是间接官能化相对于直接官能化的优点:能导入更大范围的官能团,得到种类更多的产物,为合成路线的设计增加了多样性。
Adam Noble等于2020在Nature上报道了一种通过自由基机理对C—H键进行硼化的反应[7](图6)。在390 nm LED光照和催化剂的存在下,反应给出了sp3C—H转化为硼酸酯的产物。在Fawcett等于2017年报道的脱羧硼化相关反应的基础上[8],他们试图得到一个更活泼的自由基,活泼到足以和sp3C—H反应,因此选择了三氟乙醇作为反应的试剂。
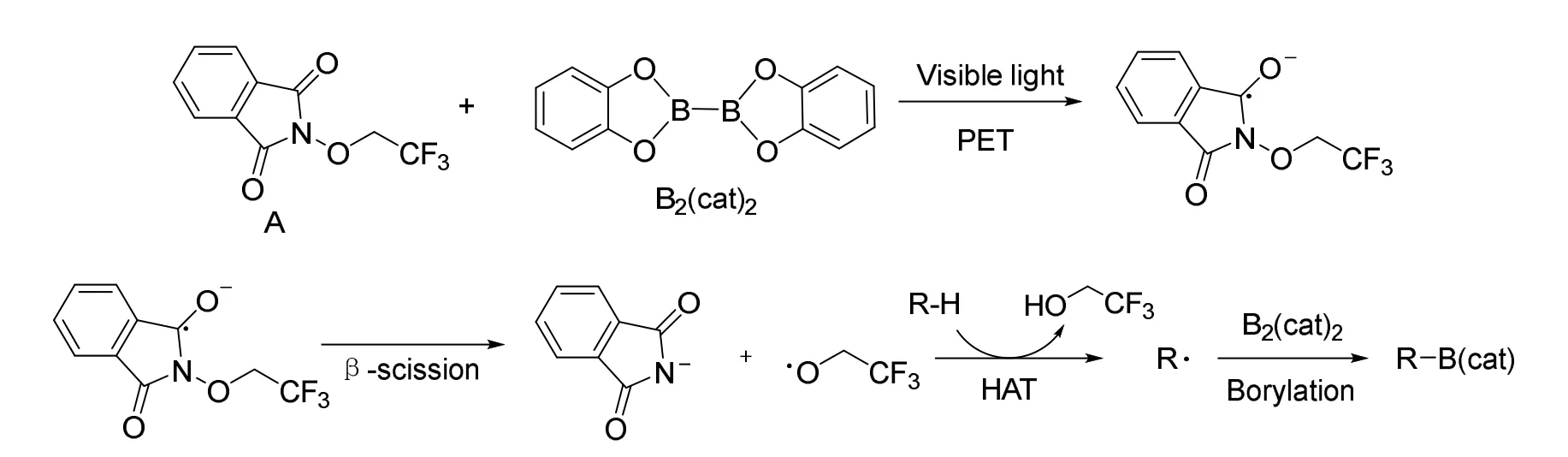
图6 自由基碳硼化反应机理
但反应的选择性却与研究人员所预料的不一致。自由基反应攫氢的选择性理论上应当和C—H键的键能大小相关,也就是说,生成的自由基相对稳定,C—H键能越小越容易被攫氢。按照这样的推测,二级碳上的H原子应当有更高的反应活性,然而事实上实验观察到此反应对甲基上的氢有着相当高的选择性,即使是在烯丙位存在H原子时,也优先对甲基进行硼化(图7)。在sp2C—H存在下同样先与sp3C—H反应,我们推测原因与产生的自由基稳定性相关。这样的选择性恰好与过渡金属催化的硼化反应相反[9],因此二者相互结合构成了一个选择性更为全面的硼化策略。类似的,对四乙基硅烷的硼化也展现出了相反的选择性,过渡金属催化受到位阻效应的影响,主要在端位甲基进行反应[10];而此反应则更倾向于在亚甲基上进行。
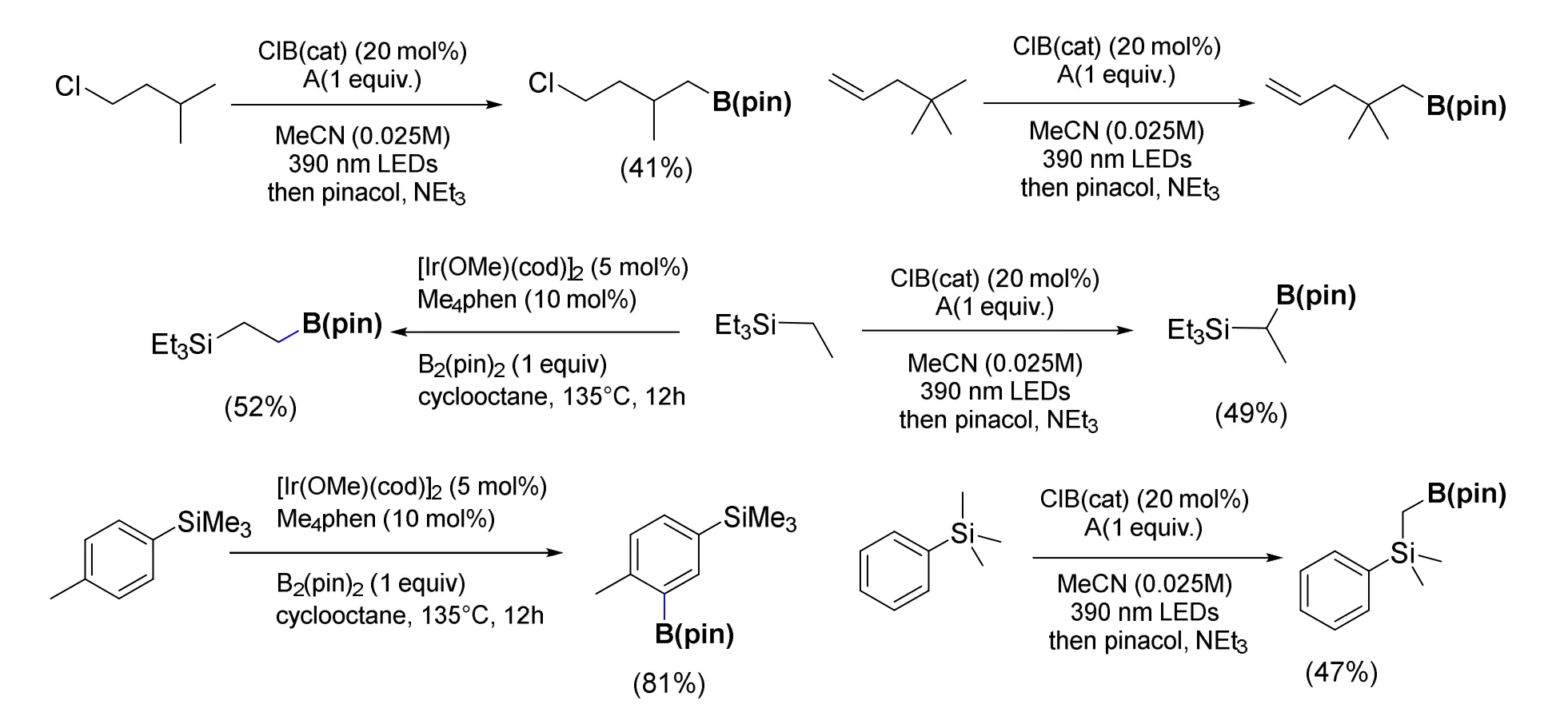
图7 C—B化的选择性对比
通过对反常的实验现象的分析,Adam Noble 课题组设置了一组改变催化剂种类的对照试验,发现氯离子的存在是该反应能否顺利进行的关键[7]。因此他们推测,反应一个关键中间体是被氯离子稳定的自由基,该自由基有一定的体积,同时不太稳定,因此该中间体优先与空间位阻小的C—H进行反应,得到甲基的C—H被硼化的产物(图8)。但是,此反应的选择性往往容易被电子效应影响。
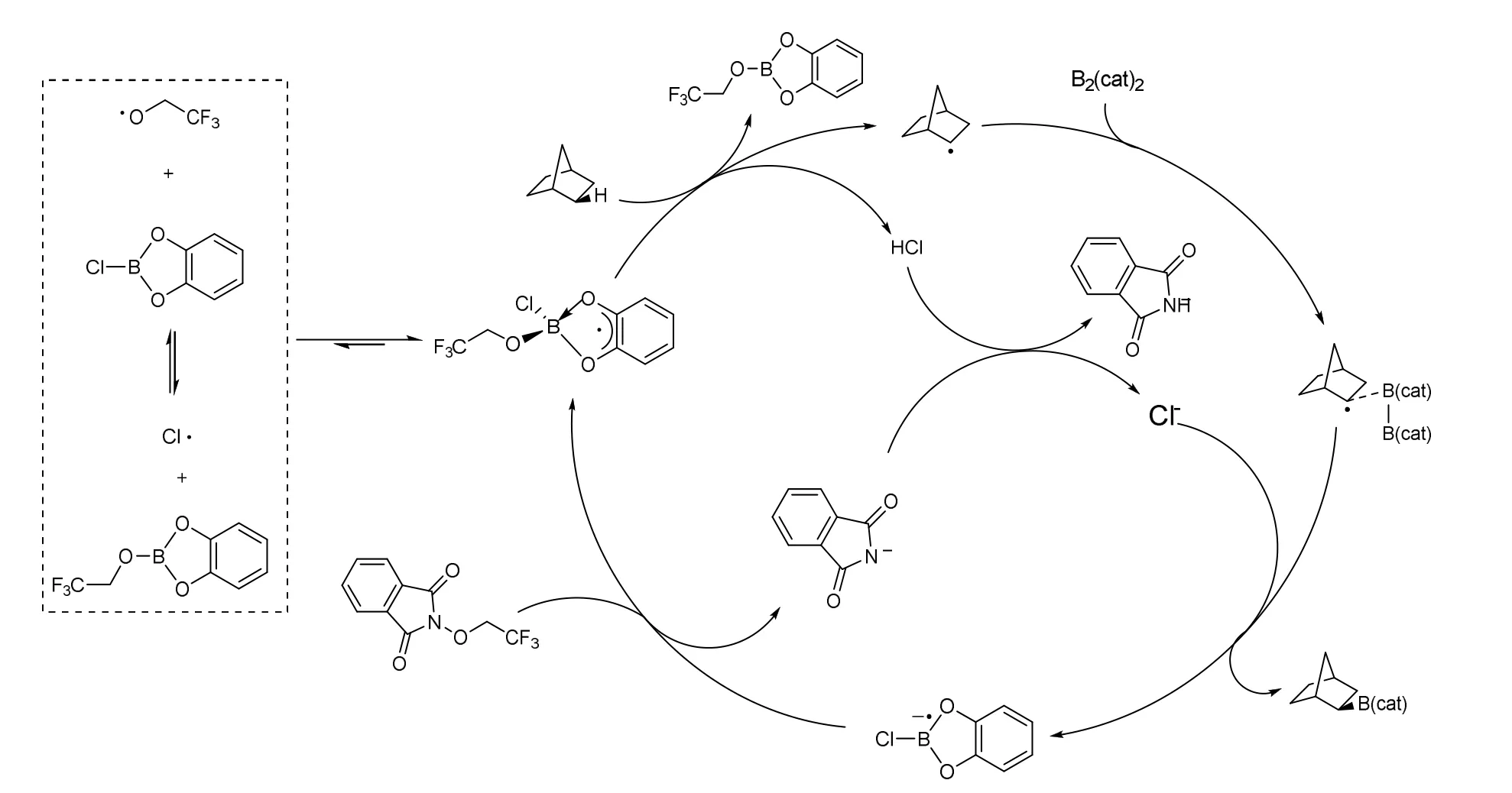
图8 碳硼化反应催化循环示意图
1.2 外围编辑在合成中的应用
以丹酚酸B (Lithospermate)的全合成为例,很明显构建两个分子片段之间的碳碳双键和氧杂的五元环系是合成的关键。常规反应要想构建碳碳双键,一般通过缩合反应或Wittig反应及其衍生反应。Jacobson等于1979年首次报道了一条通过Knoevenagel反应构建两个分子片段之间双键的路线[11](图9)。此后,Bergman等也于2005年报道了一条利用过渡金属催化的合成路线[12]。
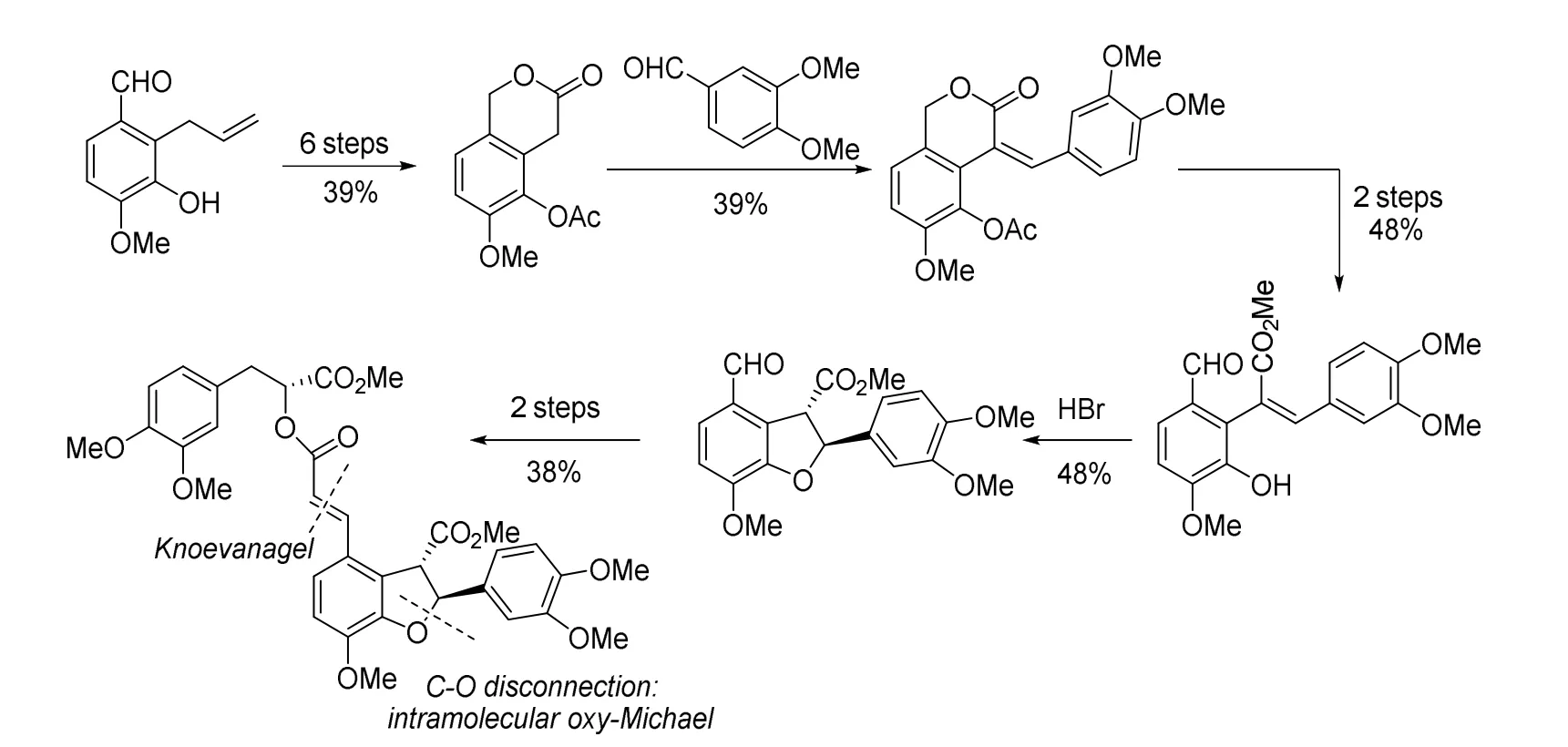
图9 Knoevenagel反应合成丹酚酸B的路线
而余金权课题组则直接通过分子间的C—H活化反应构建了两个分子片段之间的碳碳双键[13](图10),简化了实验操作,为天然产物的全合成路线设计开创了新的领域,展现了分子编辑技术的独到之处与优势。
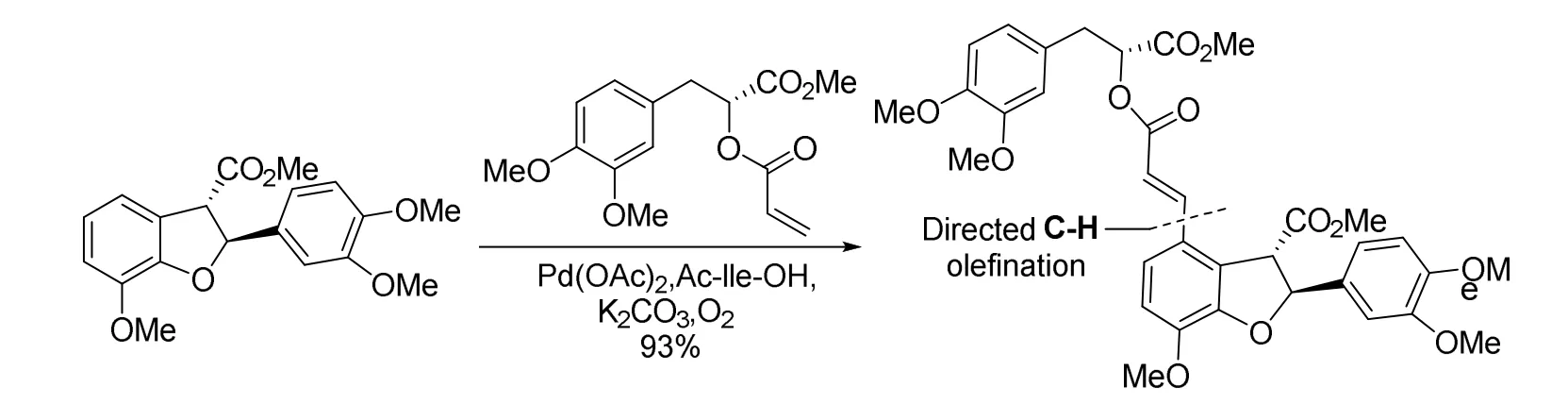
图10 直接C—H活化合成丹酚酸B的路线
2 骨架编辑(skeletal editing)
2.1 骨架编辑的主要策略
骨架编辑对底物分子在原子尺度上进行操作,其中涉及单原子(目前主要是N原子)转换的操作一般是通过与活性物种相应的等价物试剂进行反应。也有工作通过分子骨架的跃迁、重排,从而实现表面上的单原子的增删。
2.1.1 N原子的增删
以N原子的编辑为例,来自芝加哥大学的Levin教授课题组开发了一种以异头酰胺(Anomeric amide)作为氮宾等价物的骨架编辑反应[14]。首先二级胺与异头酰胺反应,离去醋酸根,形成N—N键,经过还原消除,失去一分子酯,得到异二氮烯(isodiazene)中间体。而后经过单电子转移的自由基历程,迅速失去一分子氮气,产生的碳自由基快速耦合,得到删去N原子的产物(图11)。
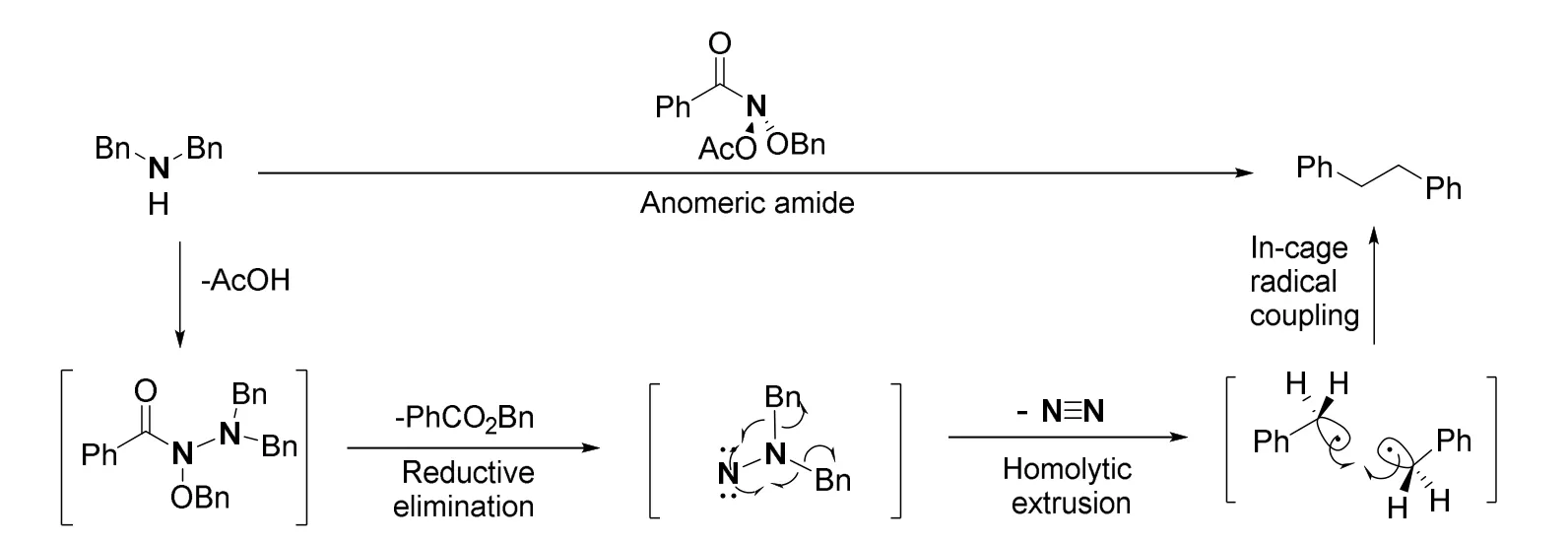
图11 异头酰胺引导氮原子删除的反应历程
Levin教授课题组对不同基团取代的异头酰胺试剂进行了探索,实验结果发现,当乙酰氧基取代时会有相当一部分N-酰基化产物生成,收率仅为35%;而当取代基更换为叔丁酰氧基时,副产物的生成受到了明显的抑制,且收率上升到57%;倘若将苯基更换为吸电子能力更强的对三氟甲基苯基,则收率可提升到74% (图12)。副产物的产生是因为酰基碳与酰胺中的氮在亲核反应一步互相竞争,很明显,如果增加酰基碳的位阻,将有利于N上的亲核取代,使副产物的生成减少。同时,N的亲电性越强,亲核反应一步就越快,同时副产物也越少。
由于自由基的高反应活性,这个反应产生的C自由基应当有稳定化因素稳定之,否则氮气将不会脱除,而是发生异二氮烯中间体的二聚(图13)。
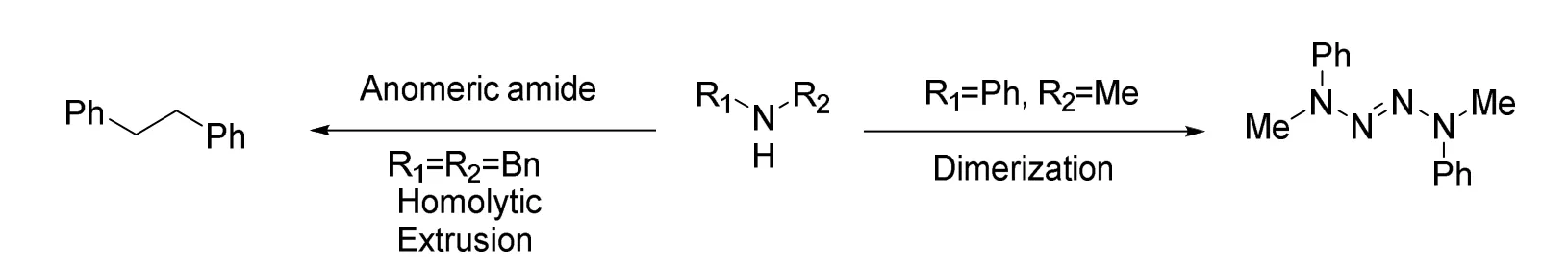
图13 缺乏自由基稳定因素导的中间体二聚示意图
Levin教授开发的这种策略有着较好的官能团兼容性,相比以前使用氧化汞和四醋酸铅氧化肼衍生物的方式,这种方式更为温和,并且不涉及繁琐的试剂,还极大地简化了合成路线的设计,一定程度上开创了新的逆合成思路。由于能和氨基进行反应,也有事实证明Anomeric amide具有使遗传物质突变的可能[15]。
除了异头酰胺,三价碘试剂也可以用于编辑骨架中的N原子(图14)。诺丁汉大学的Antonchick课题组发展了一种基于三价碘试剂进行氮原子删除的策略,同样经过了异二氮烯中间体[16]。在三价碘试剂HTIB的作用下,甲酸铵转化为碘氮宾(iodonitrene)。作为氮宾的等价物种,碘氮宾与胺反应后,得到异二氮烯,失去一分子氮气并得到分子内自由基耦合的产物。并且此反应具有高度的立体专一性。
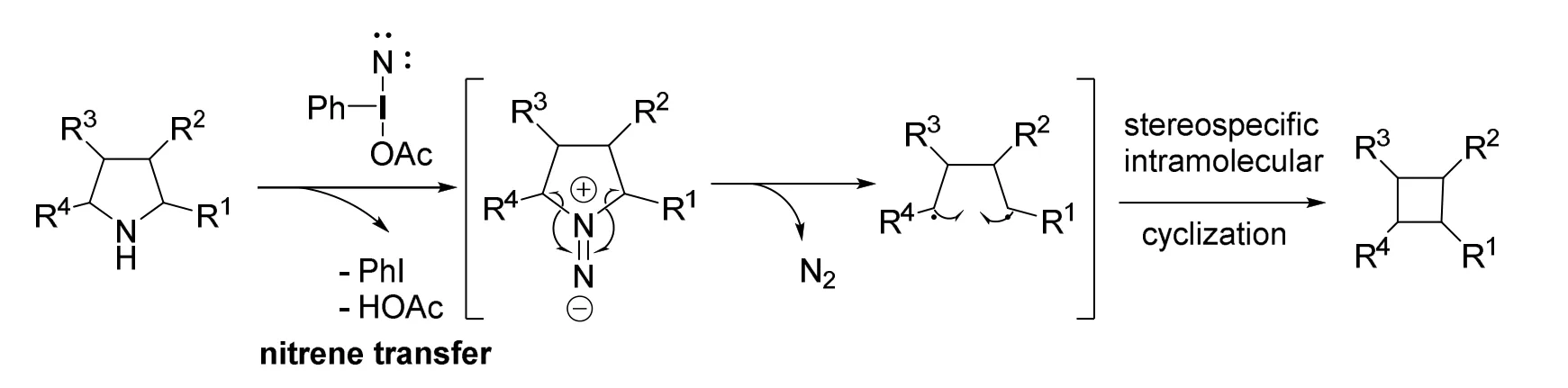
图14 碘氮宾引导的氮原子删除
这种策略大大简化了四元环的逆合成分析,为含四元环的分子的合成提供了一种新的思路,使合成思路不再拘泥于[2+2]反应和一些其他重排反应。
类似地,陆红健教授课题组利用二叠氮砜(N3SO2N3)与胺反应,通过胺磺酰叠氮中间体发生类Curtius重排也能得到异二氮烯,并发生类似的脱氮反应(图15)[17]。
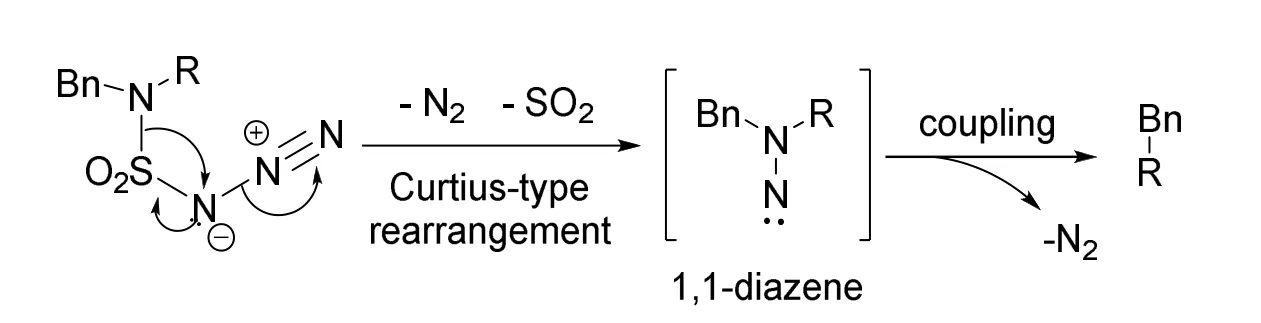
图15 胺磺酰叠氮中间体的脱氮历程
总的来说,上述几种反应删除氮原子的思路,都是利用各种试剂作为氮宾的等价合成子,与底物胺反应,构建异二氮烯中间体,最后失去氮气。在上述几种方法之外,异二氮烯中间体还可以与Angeli盐(Na2ONNO2)反应得到[18]。除了利用氮宾等价物得到异二氮烯,也可以通过胺与亚硝酸反应生成亚硝胺,还原得到肼再氧化失去氮气得到相应的产物[19],也有的工作报道了由Cl+引导,删除N原子引起酞嗪环的缩环反应[20]。
与N原子的删除不同,有机化学家们较早就探索出了可以实现N原子插入的有机反应。1979年Masanori Somei和Yoko Kurizuka便已经开发出一种向吲哚环中引入一个氮原子得到噌啉环的策略[21](图16)。将吲哚氨基化后在酸性条件下水解开环,闭环后再用硝基苯氧化,得到噌啉环。
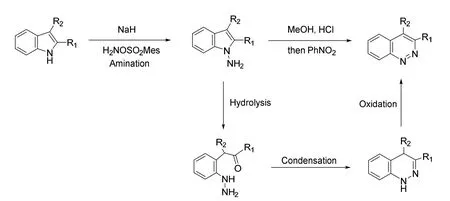
图16 氮原子插入的已有策略
同样利用碘氮宾作为氮宾的等价合成子,苏黎世联邦理工学院的Bill Morandi教授课题组开发了一种向吲哚环中插入N原子的途径[22](图17)。在保护基存在下,吲哚具有亲核性的双键优先与碘氮宾反应,形成氮杂三元环的中间体,这个中间体再开环、离去碘苯,则得到产物喹唑啉。
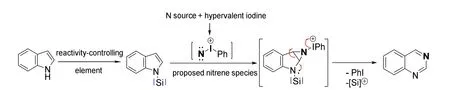
图17 碘氮宾引导的氮原子插入
由于氮原子同样会与氮宾发生反应,发生前述的脱氮反应,他们通过引入氮原子上的保护基来进行反应位点的控制,保证碘氮宾只与双键反应。实验表明,保护基种类不同会导致产率的波动(图18),我们推测这与保护基的位阻以及脱除的难易程度相关。保护基越稳定,发生的副反应越少,同时脱除的难度也变大,产率就越低。
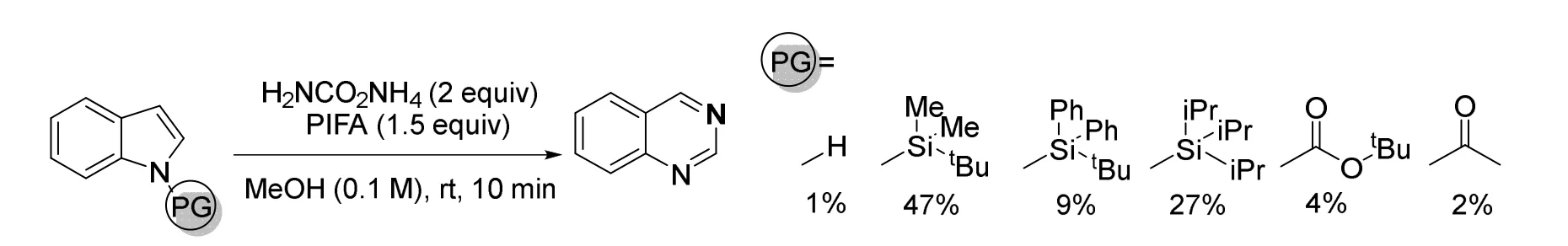
图18 不同保护基对产率的影响
事实上,由于取代方式的不同,也会得到不同位置的氮原子插入产物。如果底物是多环吲哚骨架,当吲哚的并环是五元或六元环时,得到的产物将会是喹喔啉;当环系进一步扩大为七元环时,产物又以喹唑啉为主。很明显,如果并环底物想要生成喹唑啉底物,将得到环系扭曲的结构,使氮原子共轭程度下降从而降低其稳定性;而喹喔啉骨架将维持其平面结构,使得产物能量更低。事实上,根据密度泛函理论的计算,喹喔啉骨架的产物将会比喹唑啉骨架的产物稳定70 kcal·mol-1(1 kcal·mol-1= 4.184 kJ·mol-1)之多(图19)。虽然生成喹唑啉结构的过程由于活化能更小,是动力学有利的;而生成喹喔林结构的过程由于底物更稳定,是热力学控制的。
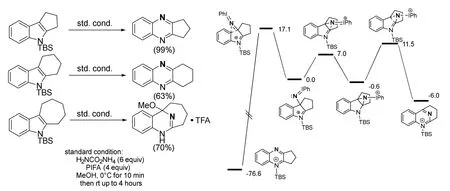
图19 不同取代方式对产物的影响以及吲哚五元并环密度泛函理论计算结果
与传统化学方法不一致的是,南京大学的程旭教授等开创了一种电化学方法向环中引进N原子[23](图20)。在石墨作正极,银作负极以及高氯酸镁作电解质的情况下,可以获得52%的收率。尽管电化学方法可能存在着电极钝化、过度反应等问题,这种策略仍体现出电化学所具有的简便性,还有着原子经济性高、副产物无毒害和官能团兼容性好的特点。
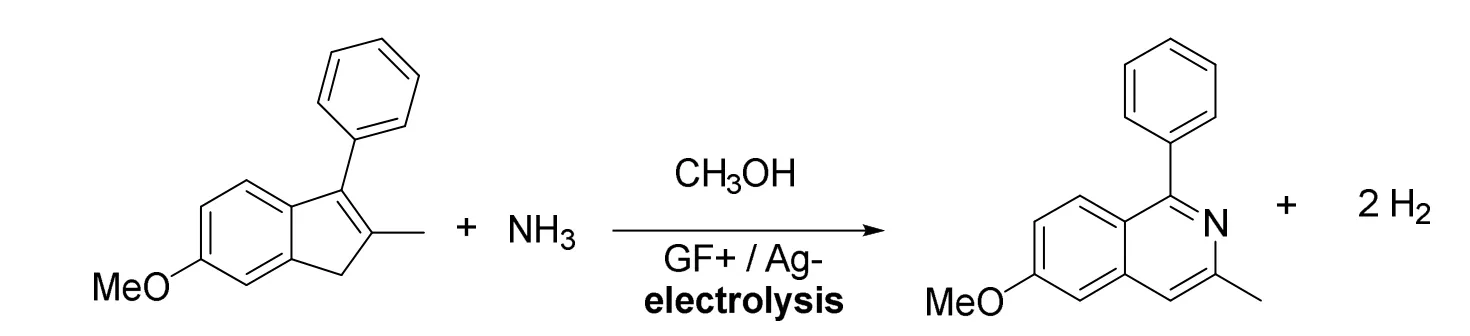
图20 电化学方法引导的氮原子插入
由于选择性的不同,上述几种向环中插入N原子的方法互相补足,成为分子骨架变动的一套新策略。
2.1.2 C原子的增删
除了电化学方法,光促的重排反应也可以引导杂环中碳原子的删除。Levin教授课题组探究了汞灯和LED照射等多种条件,发现在390 nm LED照射下,喹啉氧化物能以相当高的产率转化为六并七的杂环环系[24],该中间体可以分离[25],并可以在酸的作用下重排为吲哚的酰化物,在碱性条件下脱除酰基即可得到删去一个碳原子的吲哚产物(图21)。
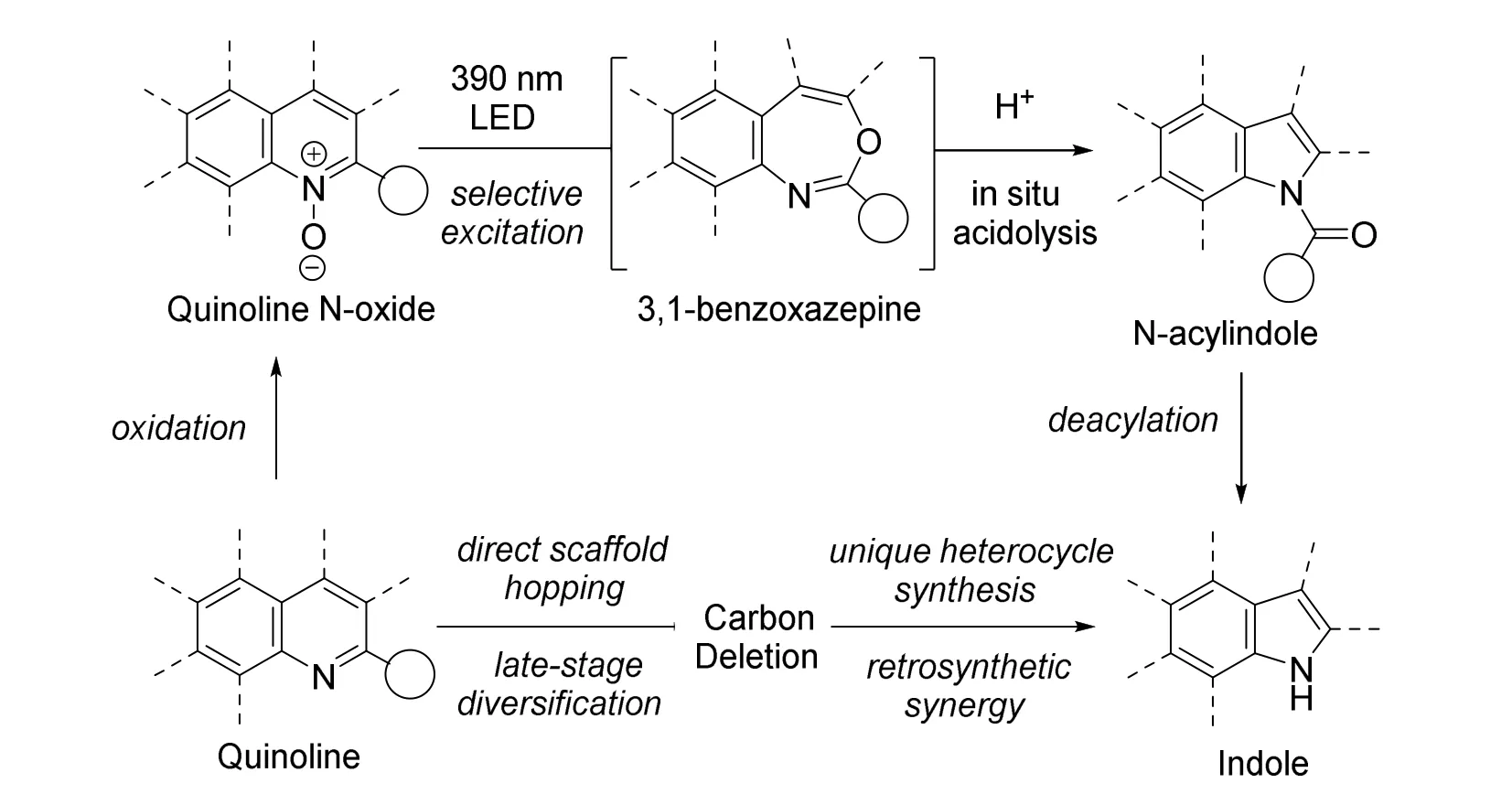
图21 光促骨架重排引导碳原子的删除
有趣的是,如果使用波长范围更广的汞灯进行照射,不仅会使产物产率降低,同时会得到许多奇怪的重排产物(图22),以及喹啉氧化物的脱氧产物。我们推测正是因为汞灯产生了紫外和可见光范围内更广的照射波长,390 nm以外其他波长的光使得产物产生了不可控的光化学反应,得到了意料之外的副产物,因此降低了产率。
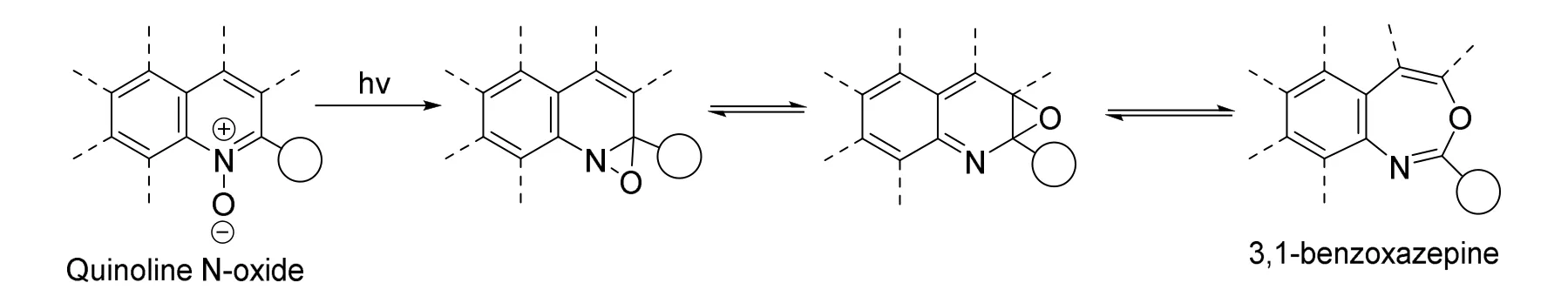
图22 骨架重排历程
此外,Levin教授课题组还在Ciamician-Dennstedt重排的基础上,开发了一种metal-free的合成策略[26]。通过氧化脒得到了可以分离的卡宾碳正离子(carbynyl cation)的等价合成子,再利用试剂与吲哚反应成功地在五元环中引入了一个碳原子(图23)。实验发现此反应的区域选择性与吡咯环的位阻有较大关系,反应往往发生在位阻更小的一侧。这样的合成策略极大地简化了药物相关化合物的设计与合成,打破了实际逆合成分析中的阻碍因素,与传统的Ciamician-Dennstedt反应相比又有着良好的官能团兼容性与更高的产率。
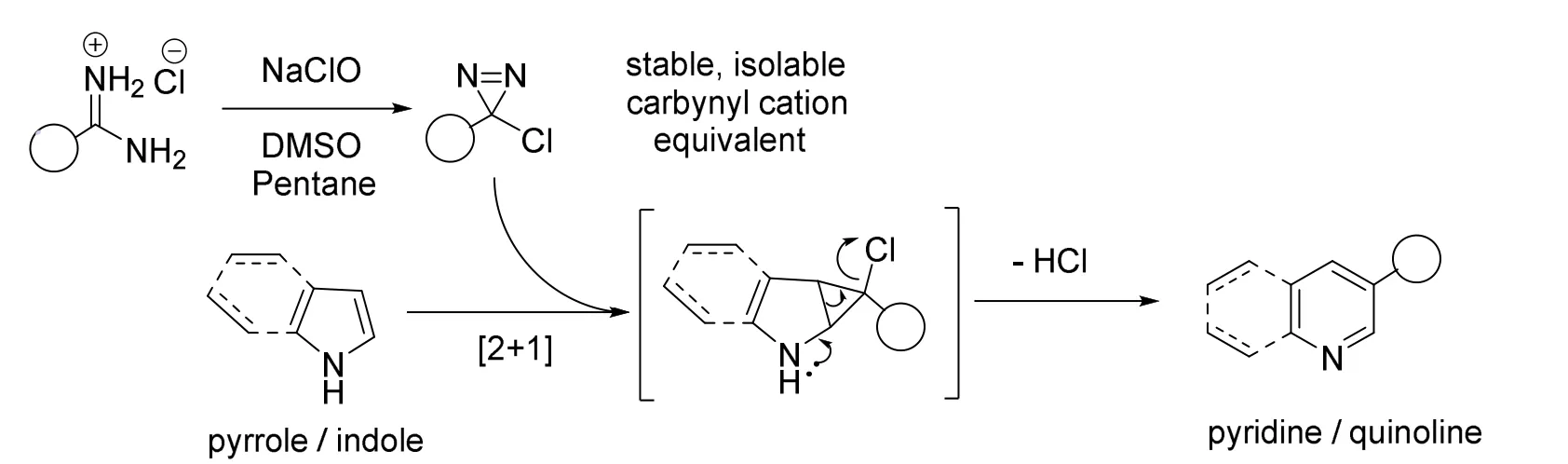
图23 碳卡宾正离子引导的增碳反应
2.1.3 氧原子的增删
经典的氧原子插入反应有如Baeyer-Villiger反应[27]和Dakin氧化[28],这两个反应都以含有过氧链的试剂作为媒介,将过氧链中一个氧原子转移到分子骨架当中去。不过这样的反应主要有以下几个不足:首先是需要在羰基的引导下才能实现氧原子的插入,适用范围不广;其次,过酸的反应活性高,过酸容易与分子内其他官能团如双键等反应,官能团兼容性差。目前有效的氧原子插入反应仍有待开发。
至于氧原子的删除,还要从醚的转金属化说起。由于已开发出的以醚作为底物进行偶联的反应只能利用醚一半的烷基[29],碳原子经济性不高,曹章杰课题组改进了这一策略。在转金属催化剂的存在下,新反应能充分利用底物醚中的烷基,而最终的反应结果是得到醚的脱氧产物[30](图24),实现了形式上氧原子的删除。
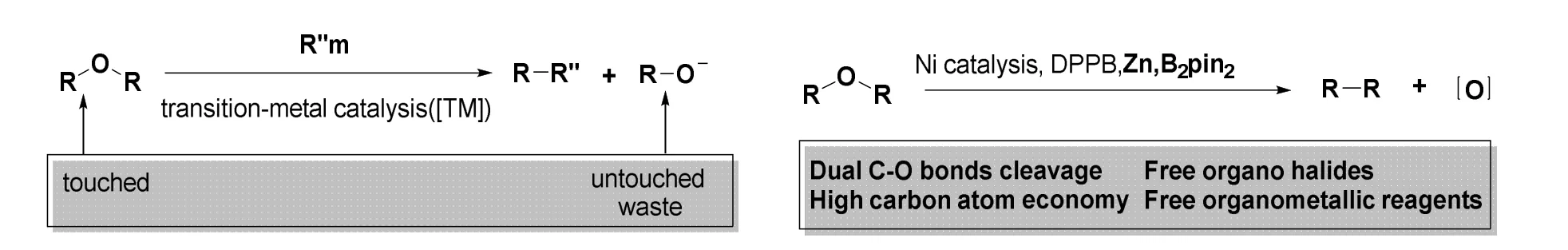
图24 镍催化的脱氧反应
这个反应中,锌和硼酸酯作为脱氧剂捕获醚中的氧原子,而锌同时作为主要的还原剂推动以镍为中心金属的催化循环(图25),最终在金属镍各个物种的转换里实现醚的脱氧反应。
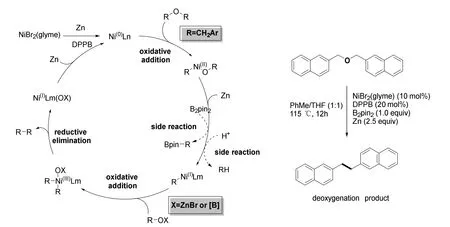
图25 脱氧反应的催化循环
这个反应目前仍有着诸多的限制,如只能用于苄醚的脱氧,且对简单的二苄醚无效。此外,如果氧的α位有烷基取代,将会发生速度更快的β消除反应,而得不到目标产物。作为试剂,锌和硼酸酯有着相互促进的作用,其中,反应如果不在锌的存在下将无法进行,而缺乏硼酸酯将导致产率大大降低。配体的种类也有着较大的影响,经过多次实验,有着更大扭角的DBBP (1,4-Bis(diphenylphosphino)butane,1,4-(双二苯基膦)丁烷)能得到更好的收率。
2.1.4 骨架跃迁
除了通过骨架跃迁实现上述单原子编辑反应,部分重排反应也可以直接引导骨架的重组与分子结构的编辑。一种光化学反应可以实现饱和杂环骨架的缩环。在400 nm LED的存在下,羰基引导的自由基裂解使哌啶环系收缩为四氢吡咯环系,同时进行了碳环上的官能化[31](图26)。这种策略的好处在于它不仅免去了试剂的使用,简化了实验操作,还同时实现了骨架编辑(对分子骨架的调整)和周围编辑(碳环周边的官能化)。
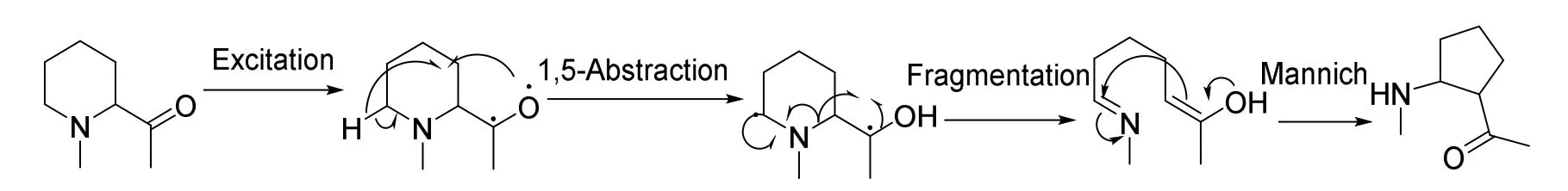
图26 光化学引导的骨架编辑历程
2.2 骨架编辑在合成中的应用
以降脂药物,HMG-CoA抑制剂氟伐他汀(Fluvastatin)为例,其IC50 (half maximal inhibitory concentration,半抑制浓度)值为8 nmol·L-1,而如果将分子核心的吲哚环改为喹啉环,并对环上进行一定程度的修饰,就得到匹伐他汀(Pitavastatin)。匹伐他汀的IC50值为5.8 nmol·L-1。通过对氟伐他汀进行一步碳原子插入反应,并修饰其分子骨架,即可得到药效更好的匹伐他汀。类似地,IC50值1.1 μmol·L-1的依托考昔(Etoricoxib)通过分子编辑反应,对环上进行碳原子的删减和氮原子的插入,可以得到IC50值50 nmol·L-1的塞来昔布(Celecoxib) (图27),后者药效更为稳定。
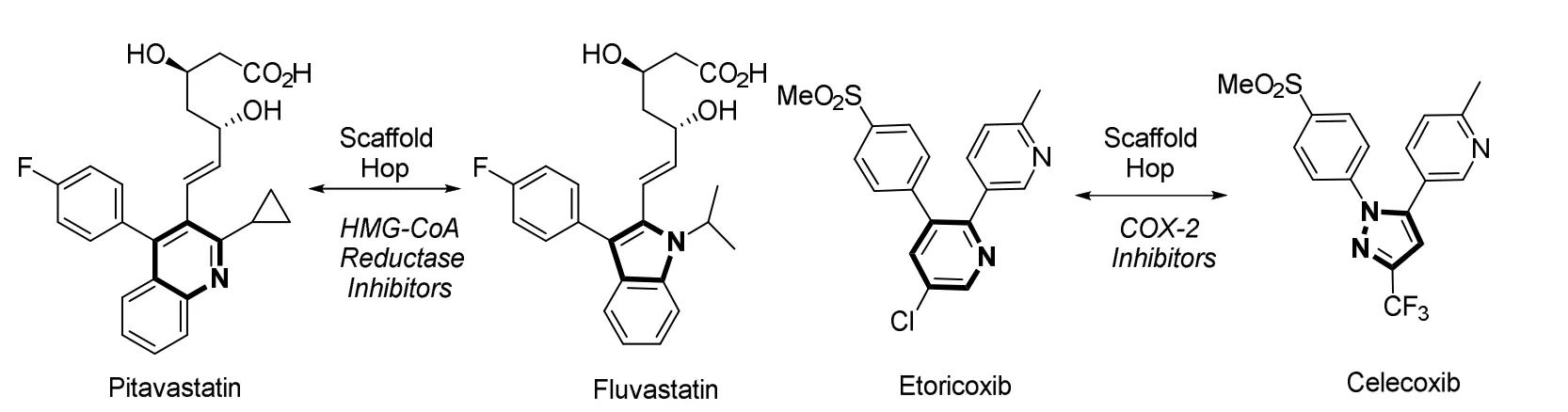
图27 匹伐他汀与氟伐他汀、依托考昔与塞来昔布之间的相互转化
除此以外,骨架编辑还在其他一系列分子的合成中有着广泛的应用。比如图28展示的通过光化学反应实现同时进行骨架编辑和外围编辑,快速得到目标分子。在第一个例子中,甘氨酸α肽通过光化学反应以29%的收率得到了甘氨酸β肽,而另一个例子中吡喃葡萄糖衍生物通过光化学反应以16%的收率得到了开环产物[31](图28)。尽管收率比较低,但考虑到部分底物的可回收性,这样的反应方便、经济而且快捷。
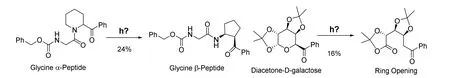
图28 利用光化学反应进行骨架编辑
结合上述例子来看,骨架编辑反应在合成中有着广泛的应用,尤其在后期的分子骨架变动(latestage scaffold hopping),部分反应还可以达到同时进行外围修饰的目的,通过较少的步骤合成中间体。在部分特殊结构的合成中(如环丁烷结构),分子编辑反应展现出得天独厚的优势,为开辟新的合成路线、设计新的策略奠定了基础。
3 对分子编辑技术的展望
综合外围编辑技术和骨架编辑技术来看,无论是已经得到一段时间发展的外围编辑,还是近年来兴起的骨架编辑技术,分子编辑技术的潜力正在不断地被挖掘,这种策略将逐渐展示出其独特之处。
外围编辑技术负责将分子进行官能化和修饰,骨架编辑可以用于后期分子骨架的调整,二者相结合使得合成化学家能够更为便利地设计分子的合成路线,为分子的逆合成分析提供了更多的可能。此外,分子编辑技术在后期修饰中所独有的选择性将为药物分子的优化和合成提供极大的便利!这为更有效药物的合成与设计奠定了基础。
尽管有的分子编辑反应产率尚低,骨架编辑反应还没有形成完整的体系,尽管化学家们目前还不能在杂环的任意一个位点插入任意的原子,但是分子编辑反应仍展现出巨大的潜力,仍有着很多未知的反应等待我们探索。只要我们不断地探索、完善这一系列反应,相信总有一天化学家手中的分子编辑工具箱将会完善,产率将会不断提高,试剂将会不断改进,分子编辑技术将会得到质的飞跃与发展。
这将是化学领域一次新革命的开端,相信等到分子编辑技术成熟之时,我们真的可以按照自己所想,随心所欲地设计和合成分子。借用Mark D. Levin教授的一句话来展示我们对分子编辑反应的期望,“尽管我们还有很多事情做不到,但在将来,在你的烧瓶中将苯的一个碳原子替换为氮原子,应该像使用铅笔和橡皮一样简单[32]。”
猜你喜欢
中国药学药品知识仓库(2022年10期)2022-05-29 02:59:32
电子乐园·上旬刊(2022年5期)2022-04-09 22:18:32
汕头大学学报(自然科学版)(2020年4期)2020-12-14 07:04:58
中国新技术新产品(2020年5期)2020-05-06 03:36:28
天津农学院学报(2016年2期)2016-12-01 05:40:05
信息记录材料(2016年4期)2016-03-11 15:22:36
中国塑料(2015年8期)2015-10-14 01:10:53
中国煤层气(2014年3期)2014-08-07 03:07:45
无机化学学报(2014年12期)2014-02-28 17:34:01
华东理工大学学报(自然科学版)(2014年6期)2014-02-27 13:49:40
