
CaO-Ca3Al2O6@Ni-SiO2复合催化剂制备及制氢性能
2023-01-04 09:23:12荆洁颖李文英
燃料化学学报 2022年12期
许 凯 ,刘 璐 ,荆洁颖 ,冯 杰 ,李文英
(太原理工大学 省部共建煤基能源清洁高效利用国家重点实验室, 山西 太原 030024)
1994年Han等[1]提出吸附强化CH4/H2O重整(Sorption-enhanced steam methane reforming,SESMR)制氢的概念,其主要思想是通过引入CO2吸附剂,原位移除反应产生的CO2,提高CH4转化率和产物中H2的选择性,实现一步法制取高浓度H2。这项技术明显地简化了工艺流程[2-4],降低了能耗[5]。但该技术所使用的催化剂和吸附剂在循环制氢过程中会发生烧结,造成催化和吸附性能的下降,严重影响制取H2的浓度。因此,如何制备具有高循环稳定性的双功能复合催化剂是该技术工业化需要攻克的关键问题之一。
复合催化剂具有更短的传质距离和更小的传质阻力[6],更有利于CO2的原位吸附,但随着反应的进行,复合催化剂的吸附组分和活性组分发生烧结,同时循环再生过程中复合催化剂的吸附组分经历从CaO到CaCO3再到CaO的循环往复变化,会造成复合催化剂结构的坍塌,同时活性组分Ni也会被反应生成的CaCO3包埋,引起催化性能的下降,严重影响复合催化剂的制氢性能[7]。研究人员尝试通过改变复合催化剂中的结构来解决结构坍塌的问题,Jing等[8,9]制备出具有层状结构的Ni/CaO-Al2O3复合催化剂,并探究了Ca/Al物质的量比和焙烧温度对复合催化剂的结构和性能的影响。结果表明,当Ca/Al物质的量比为3,焙烧温度为700 ℃时,制备的复合催化剂在10次循环后可以得到97%的H2浓度。但这种复合催化剂仍不能完全解决复合催化剂在循环反应中的结构坍塌问题。而核壳结构复合催化剂由于其独特的限域效应可被用于解决上述问题,其壳层的限域空间可提供足够大的空间来保证吸附组分的体积变化不会使催化剂结构坍塌,同时避免金属粒子的团聚烧结[10]。Chen等[11]用两步溶胶-凝胶法制备了以吸附剂CaO-Ca9Al6O18为核、以催化剂Ni/Ca5Al6O14为壳的双功能核壳型催化剂,该催化剂在600-650 ℃,H2O/CH4物质的量比为3.0-4.0的条件下展现出较好的制氢性能(> 93%),这归因于惰性物质的引入和催化剂壳层载体的骨架作用,但由于复合催化剂中的惰性组分含量较高(67%),导致复合催化剂的CO2吸附容量很低(约2.2 mmol/g)。Xu等[12]通过采用尿素水解法结合吸附相反应技术,成功制备以NiO为核、CaO为壳的核壳结构NiO@TiO2-CaO/Al2O3,并考察了它的制氢性能,发现该催化剂经多次循环后H2维持在90%以上,CH4转化率保持在85%,这主要是因为核壳结构对催化剂起到了支撑作用,防止了因载体烧结而导致的催化剂活性中心失活,提高了催化剂的稳定性。然而,目前所报道的核壳结构催化剂中惰性组分虽然起到了很好的支撑作用,但因其在复合催化剂中占比很大(60%-80%)导致CaO利用率不高;另外,虽然吸附组分为核的复合催化剂性能更佳,但吸附组分在反应-再生循环过程中会经历从CaO到CaCO3再到CaO的循环往复变化,很容易造成复合催化剂结构的坍塌,所谓核壳结构催化剂也变成了催化组分、吸附组分和惰性组分的混合体。因此,如何克服吸附组分在反应-再生循环过程中体积反复膨胀收缩带来的孔结构坍塌和催化组分的烧结、维持核壳结构复合催化剂的稳定性是亟待解决的问题。
本研究利用阳离子表面活性剂辅助刻蚀的机理采用自模板法设计制备了具有核壳结构的CaOCa3Al2O6@Ni-SiO2复合催化剂,考察了其吸附强化制氢性能,并对其构效关系进行了研究。所得结果对于进一步改善复合催化剂的活性稳定性具有重要的理论指导意义。
1 实验部分
1.1 实验原料
硝酸铝(Al(NO3)3·9H2O)从麦克林(Macklin)购买,硝酸镍(Ni(NO3)2·6H2O)、硝酸钙(Ca(NO3)2·4H2O)从伊诺凯(Innochem)购买,柠檬酸(C6H8O7·H2O)、正硅酸乙酯(TEOS),十六烷基三甲基溴化铵(CTAB)、无水碳酸钠(Na2CO3)、氨水(含氨 25%-28%)、乙醇(纯度 99%)从科密欧(Kermel)购买。
1.2 复合催化剂的制备
1.2.1 吸附组分CaO-Ca3Al2O6的制备
使用溶胶凝胶法制备吸附组分CaO-Ca3Al2O6[13]。具体步骤简述如下:称量3.67 g Al(NO3)3·9H2O与18.97 g Ca(NO3)2·4H2O溶于 34 mL超纯水中,称量18.94 g 柠檬酸溶于30 mL超纯水中,待全部溶解后,将两者混合,密封搅拌1 h,然后将混合溶液在80 ℃搅拌6 h,待形成凝胶后放于烘箱干燥(100 ℃干燥 10 h,120 ℃ 干燥 10 h,130 ℃ 干燥 5 h),最后将形成的干凝胶在 900 ℃(1 ℃/min)焙烧 1.5 h,得到吸附组分CaO-Ca3Al2O6。将得到的吸附组分置于管式炉中在50 mL/min CO2气氛下升温至600 ℃,然后恒温保持4 h,使吸附组分完全碳酸化,以备后续使用。
1.2.2 复合催化剂的制备
称取5.5 g完全碳酸化的CaO-Ca3Al2O6分散于160 mL乙醇中,加入36 mL超纯水和4 mL氨水,搅拌15 min,加入300 mg表面活性剂CTAB,待其完全分散后,缓慢滴加4 mL TEOS,之后将混合乳浊液在30 ℃水解3 h,以保证TEOS完全水解,然后抽滤得到滤渣,放于烘箱中60 ℃干燥过夜得到中间产物A。称取一定量中间产物A分散于200 mL超纯水中,加入181 mg CTAB和1.26 gNa2CO3,搅拌均匀后在60 ℃刻蚀5 h,抽滤得到滤渣后放于烘箱60 ℃干燥过夜得到中间产物B。将中间产物B溶于200 mL超纯水中,加入181 mg CTAB 和 2 g Ni(NO3)2·6H2O,搅拌分散后在 60 ℃搅拌5 h,抽滤得到滤渣后放于烘箱60 ℃干燥过夜,最后将干燥后的样品在750 ℃焙烧30 min得到复合催化剂CaO-Ca3Al2O6@Ni-SiO2。同时使用浸渍法制备出不含SiO2的复合催化剂作为空白对照,记为 Ni/CaO-Ca3Al2O6。
1.3 复合催化剂的表征
采用日本理学X射线衍射仪(XRD,Rigaku D/Max -3B型)分析所制备复合催化剂的晶相结构。采用化学吸附仪(AutoChem II 2920)分析复合催化剂中催化组分的还原性能。取50 mg样品放入U型管中,在300 ℃、Ar气氛下预处理1 h,待冷却至100 ℃时将气氛切换为10%H2/Ar,然后以10 ℃/min的升温速率升至900 ℃,记录TCD的信号值。
1.4 复合催化剂的CO2吸附性能
CO2吸附性能主要基于德国耐驰公司STA449F5型热重上测得的CO2吸附-脱附的数据进行计算得到,以还原后的复合催化剂作为样品,通过复合催化剂的CO2吸附容量和CaO利用率两个指标进行评估,CO2吸附容量反应了单位质量的催化剂吸附CO2量的多少,计算公式为式(1)。

式中,N为催化剂的CO2吸附容量,mmol/g; mad为吸附CO2时催化剂增重的质量,mg; MCO2为CO2的摩尔质量,g/mol; mcat为复合催化剂的质量,mg。
CaO利用率反应了一定时间内反应的CaO与总CaO的摩尔量的比值,计算公式为式(2)。

式中,X为合催化剂中CaO利用率,%;x为复合催化剂中CaO的质量分数,%; Nθ为CaO的CO2理论吸附容量,为17.86 mmol/g。
1.5 吸附强化CH4/H2O重整制氢活性评价
SESMR反应活性评价在自制固定床石英管反应器上进行。称取4 g复合催化剂(粒径0.25-0.43 mm)放于石英管恒温区,反应前,将复合催化剂前驱体在H2气氛下700 ℃还原60 min,还原完毕后,在50 mL/min的N2气氛下降温至600 ℃,然后将N2切换为CH4和水蒸气进行反应(H2O/CH4物质的量比为4.8),反应完毕后,切换为N2(100 mL/min)气氛,同时将反应温度升至750 ℃再生60 min,然后停止加热,待反应温度降至600 ℃,再进行反应。反应后的气体由海欣气相色谱仪GC-950进行检测。
2 结果与讨论
2.1 复合催化剂的制备原理
模板法是中空微球制备中应用最为广泛的方法,根据所用模板的不同,模板法一般分为硬模板法、软模板法和自模板法[14]。硬模板法制备出的中空结构一般具有相对固定的形状,而且易于控制中空结构的大小,但其模板去除过程可能会导致壳层的破裂。软模板法通常以嵌段共聚物或表面活性剂为模板,操作简单而且容易形成中空结构,但是这种方法使用大量的表面活性剂,完全去除困难。自模板法基于物质迁移速率的差异实现中空结构的制备,该方法工艺简单,成本低,而且不需要引入其他物质,模板去除也更为方便,适用于大规模生产。
本研究首先采用自模板法制备核壳结构,然后通过选择性刻蚀的方法制备中空结构的吸附组分,选择性刻蚀的机理是由于正硅酸乙酯(TEOS)水解缩合生成的SiO2内外层聚合度的不同,导致内外层SiO2对刻蚀剂的稳定性不同,从而使得刻蚀过程从内到外的进行。在SiO2形成过程中,TEOS先水解产生具有1-4个羟基的单体,这些单体缩合形成SiO2球,而前期TEOS水解缓慢,使得溶液体系中具有较多的乙氧基,这些乙氧基使内部的SiO2具有更大的孔隙率,而外部的SiO2则是由硅酸的缩合形成,这一部分的SiO2相对更致密,这就使得内部低聚集度的SiO2可以被刻蚀掉,从而形成中空结构[15]。
复合催化剂的形成过程如图1所示。首先,将吸附组分和表面活性剂(CTAB)分散在乙醇中,CTAB在范德华力的作用下吸附在吸附组分表面,同时使得吸附组分分散的更加均匀;然后加入TEOS,使其在碱性条件下水解,水解生成的SiO2表面带负电,而CTA+离子使吸附组分表面带有大量的正电荷,两种电荷间的静电相互作用使TEOS水解生成的SiO2包覆在吸附组分表面,形成一层SiO2壳;接着加入Na2CO3进行刻蚀,刻蚀掉内部SiO2后形成的硅酸盐物种在60 ℃下再次水解,然后在CTAB的作用下沉积在吸附组分表面形成新壳层,同时在吸附组分和壳层SiO2间形成空腔[16-18];最后加入Ni(NO3)2·6H2O,在壳层上面负载Ni活性组分,通过焙烧将CaCO3分解为CaO,同时除去表面活性剂,使制备的壳层具有丰富的孔道结构。
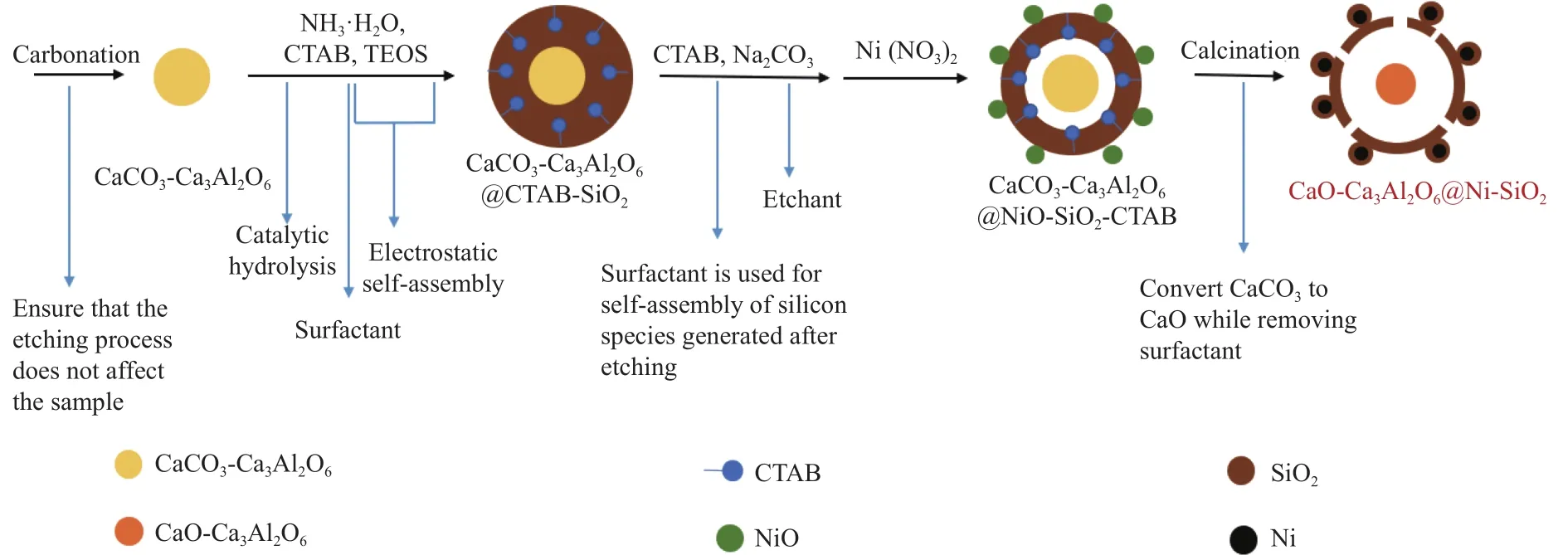
图1 复合催化剂CaO-Ca3Al2O6@Ni-SiO2的形成过程Figure 1 Formation process of composite catalyst CaO-Ca3Al2O6@Ni-SiO2
2.2 复合催化剂的性能评价
2.2.1 CO2吸附性能
复合催化剂的CO2吸附性能直接影响SESMR反应的制氢浓度,采用热重对复合催化剂的CO2吸附容量进行了分析,结果如图2所示。由图2可知,单纯吸附组分CaO-Ca3Al2O6的CO2吸附容量为9.9 mmol/g,包覆SiO2后吸附组分CaO-Ca3Al2O6@SiO2的CO2吸附容量为4.2 mmol/g,浸渍催化组分Ni后CaO-Ca3Al2O6@Ni-SiO2复合催化剂的CO2吸附容量为3.8 mmol/g,显然,制备过程中引入SiO2作为惰性组分之后,吸附组分的CO2吸附容量会下降,但并不会影响吸附组分的稳定性。对比CaO-Ca3Al2O6@SiO2和CaO-Ca3Al2O6@Ni-SiO2两者的吸附容量可以发现,浸渍Ni并不会影响吸附组分的CO2吸附容量及循环稳定性。
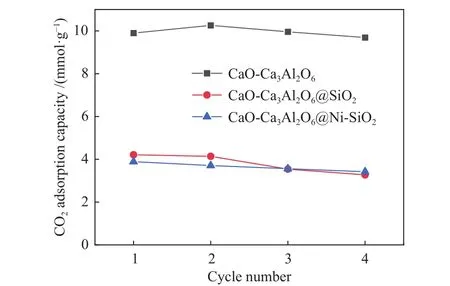
图2 SiO2对复合催化剂CO2吸附性能的影响Figure 2 Effect of SiO2 on CO2 adsorption performance of composite catalyst
2.2.2 制氢性能评价
在固定床上进行SESMR反应,具体反应条件为:700 ℃ 还原(50 mL/min H2气氛),600 ℃ 反应,750 ℃再生(100 mL/min N2气氛),催化剂装填量:4 g;CH4流量:14 mL/min;H2O/CH4物质的量比:4.8;空速:968 h-1。复合催化剂 CaO-Ca3Al2O6@Ni-SiO2的制氢产物分布如图3所示。从图3中可以看出,该复合催化剂的吸附强化时间约为25 min,在吸附强化阶段H2含量和CH4转化率可达到99%,反应60 min后,复合催化剂中的CaO吸附饱和,其重整产物中H2含量下降,CH4转化率也随之下降,直至理论计算值。10次循环后催化剂依旧保持着较高的制氢性能,其产物中H2含量仍能保持在97.3%(相比于第一次下降约2.3%),CH4转化率保持在91.8%(相比于第一次下降约7.7%)。可以看出10次循环后,CH4转化率相比于出口H2含量的下降程度更大,故推测出现这种情况的原因可能源于复合催化剂中催化组分Ni催化性能的下降。
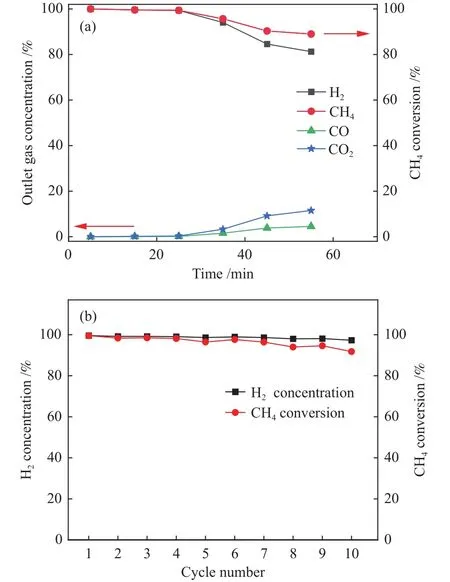
图3 单次反应和10次反应的各产物含量和CH4转化率Figure 3 Concentration of each product and CH4 conversion in the first and tenth reactions
为了探究复合催化剂中SiO2的作用,采用同样的方法制备了Ni/CaO-Ca3Al2O6(即制备方法一样,但不添加TEOS)作为空白对照,然后将其应用于SESMR反应,其结果如图4所示。由图4可知,Ni/CaO-Ca3Al2O6制氢效果较差,其产气中H2含量仅为16%,此时CH4转化率仅为5%,这可能是因为制备催化剂时750 ℃焙烧使CaCO3分解成CaO的过程中体积变化导致了催化剂结构坍塌,使得Ni被覆盖,导致催化活性降低。说明SiO2在复合催化剂中起到了结构稳定的作用,使得复合催化剂在反应过程中不会因为吸附组分体积膨胀而导致结构坍塌,同时也保护了活性组分Ni不会包埋。

图4 Ni/ CaO-Ca3Al2O6重整反应中各产物含量和CH4转化率Figure 4 Concentration of each product and CH4 conversion in the reforming reaction of Ni/ CaO-Ca3Al2O6
进一步将本研究所制备CaO-Ca3Al2O6@Ni-SiO2催化剂与文献中报道复合催化剂的制氢性能进行了对比,结果如表1所示。从表1中可以看出,相比催化剂和吸附剂的简单机械混合[19],表1中所有复合催化剂均具有较好的制氢效果,在相似条件下,CH4转化率和出口H2含量分别保持在82%-97%和85%-96%,但即便组成相同的催化剂,其制氢性能也有明显差异,说明复合催化剂的结构也极大影响了催化剂的性能。

表1 不同复合催化剂的制氢性能Table 1 Hydrogen production performance of different composite catalysts
相比文献报道的复合催化剂,本研究制备的CaO-Ca3Al2O6@Ni-SiO2催化剂由于其独特的设计理念和制备方法,使得制备的催化剂在反应时的体积膨胀和收缩都在SiO2壳层内进行,不会造成催化剂结构的坍塌,极大提高了催化剂的循环稳定性,不仅在初次反应时就达到了99.6%的制氢效果,在10次循环后仍保持着97.3%的制氢含量。但考虑到10次反应后的CH4转化率下降,复合催化剂中催化组分的抗烧结性能有待进一步提高。
2.3 复合催化剂结构
为了进一步确认所制备复合催化剂的结构并探究复合催化剂高制氢性能和CH4转化率下降的原因,采用ICP-OES、XRD、SEM以及TEM等表征手段对复合催化剂的组成和结构进行了更深入的分析。
2.3.1 组成分析
采用ICP-OES对复合催化剂的元素组成进行测定,其结果如表2所示。由表2中数据可知,复合催化剂中主要含有Ni、Ca、Al、Si、O五种元素,对应Ni、CaO、Ca3Al2O6、SiO2四种物质。其中,CaO含量约为66.84%,其理论吸附容量为11.9 mmol/g;惰性组分Ca3Al2O6含量为19.98%,明显低于文献中报道的67%[11]。复合催化剂中的惰性组分常用来稳定复合催化剂的结构,同时分隔开活性位点,减少CaO的烧结,但过高的惰性组分会使得复合催化剂CO2吸附容量的下降,同时增大CO2在催化剂颗粒中的传质阻力,影响复合催化剂的吸附性能。本研究制备的CaO-Ca3Al2O6@Ni-SiO2复合催化剂在较低的惰性组分含量(19.98%)下保证了复合催化剂的CO2吸附性能和循环稳定性,避免了因惰性组分添加而导致CO2吸附容量的大幅下降,并在10次SESMR反应中保持较高的制氢性能(> 97.3%)。

表2 复合催化剂CaO-Ca3Al2O6@Ni-SiO2的组成Table 2 Composition of composite catalyst CaOCa3Al2O6@Ni-SiO2
2.3.2 晶相结构分析
采用XRD对反应前后的复合催化剂的晶相结构进行分析,结果如图5所示。由图5可知,制备的复合催化剂具有CaO、Ni、Ca3Al2O6的特征峰,说明制备过程并没有对吸附组分CaO-Ca3Al2O6造成影响,没有引入新的杂质,制备的复合催化剂仍然保持其原有特征。而反应前后两者都具有Ca(OH)2的特征峰,这可能是因为复合催化剂中的CaO吸收了空气中的水,反应后的催化剂多了CaCO3的特征峰,说明再生过程并没有使CaCO3完全分解。但是反应前后均没有在2θ为23°处发现无定形SiO2的大包峰,这可能是因为水解生成的SiO2经过后续的刻蚀和焙烧过程,使得最终包覆在吸附组分上的SiO2分布均匀,无法被检测到。通过谢乐公式对反应前后Ni粒径进行估算,发现反应后Ni粒径由18.9 nm增至21.0 nm,进一步证明了复合催化剂10次循环后CH4转化率下降的原因是催化组分发生烧结。
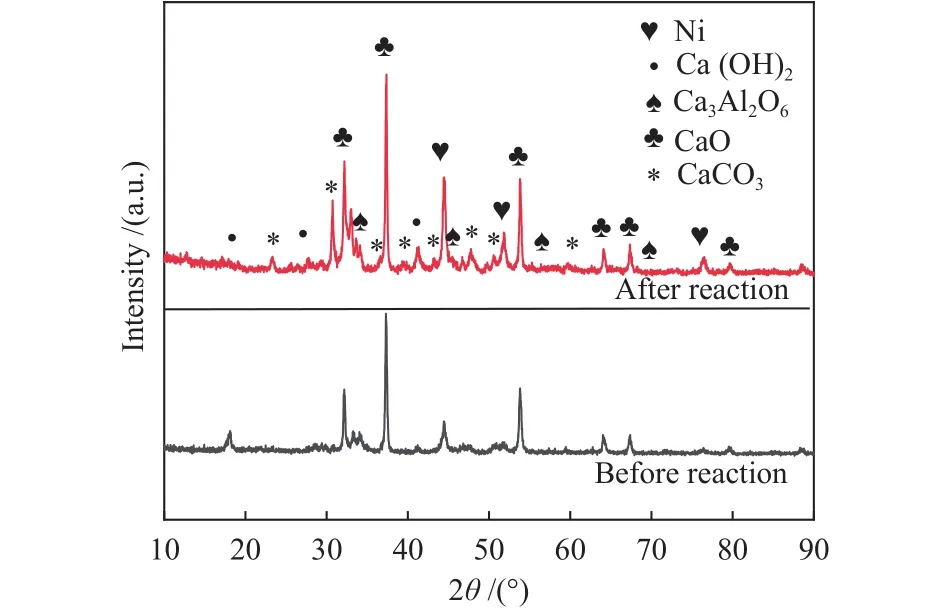
图5 反应前后复合催化剂的XRD谱图Figure 5 XRD patterns of the composite catalyst before and after the reaction
2.3.3 还原性分析
采用化学吸附仪考察复合催化剂中催化组分的还原性能,其中,CaO-Ca3Al2O6吸附组分和CaOCa3Al2O6@Ni-SiO2复合催化剂的H2-TPR谱图如图6(a)所示。从图6(a)中可以看出,CaO-Ca3Al2O6在571 ℃出现了一个特征峰,该位置的峰并非金属氧化物的还原峰,而是CaCO3分解产生的CO2造成的峰。而CaO-Ca3Al2O6@Ni-SiO2复合催化剂在480、610和855 ℃出现了三个不同的特征峰,对比CaO-Ca3Al2O6的H2-TPR谱图可以看出,480和855 ℃下对应的特征峰为NiO的还原峰,而610 ℃对应的峰为CaO-Ca3Al2O6吸附组分在H2气氛下发生反应而导致的特征峰,相比单独的CaOCa3Al2O6吸附组分而言,该特征峰的位置由571 ℃移动至610 ℃,这可能是因为SiO2包覆在吸附组分表面,造成了吸附组分中CaCO3分解温度的升高。为进一步确认610 ℃对应特征峰的归属,将复合催化剂在600 ℃ Ar气氛下进行热预处理,然后在化学吸附仪上考察了复合催化剂的还原性,其结果如图6(b)所示。由图6(b)可知,600 ℃预处理后的复合催化剂只有两个特征峰,说明610 ℃对应的特征峰并非NiO的还原峰。为进一步确定610 ℃温度下出现特征峰的原因,在热重上对复合催化剂的失重情况进行了分析,如图7所示。由图7可以看出,复合催化剂在350和600 ℃出现明显失重现象,结合复合催化剂的组成可知,在350 ℃时质量下降源于复合催化剂表面吸附Ca(OH)2的分解,而600 ℃时的失重源是CaCO3,故可判定图6(a)中610 ℃对应的特征峰为CaCO3分解生成CO2与H2反应的消耗峰。Hu等[29]的研究也发现,对于Ni-Ca基双功能复合催化剂而言,在610 ℃对应的峰为H2与CO2反应的消耗峰。说明复合催化剂具有两种不同形式的Ni物种,这两种Ni物种与载体间的相互作用力不同,即催化组分Ni在复合催化剂中有两种不同的存在形式。在480 ℃对应的Ni与SiO2之间具有较弱的相互作用力,属于负载在外表面的Ni,而855 ℃对应的Ni与SiO2之间具有较强的相互作用力,对应于Ni@SiO2的包覆型结构的Ni。
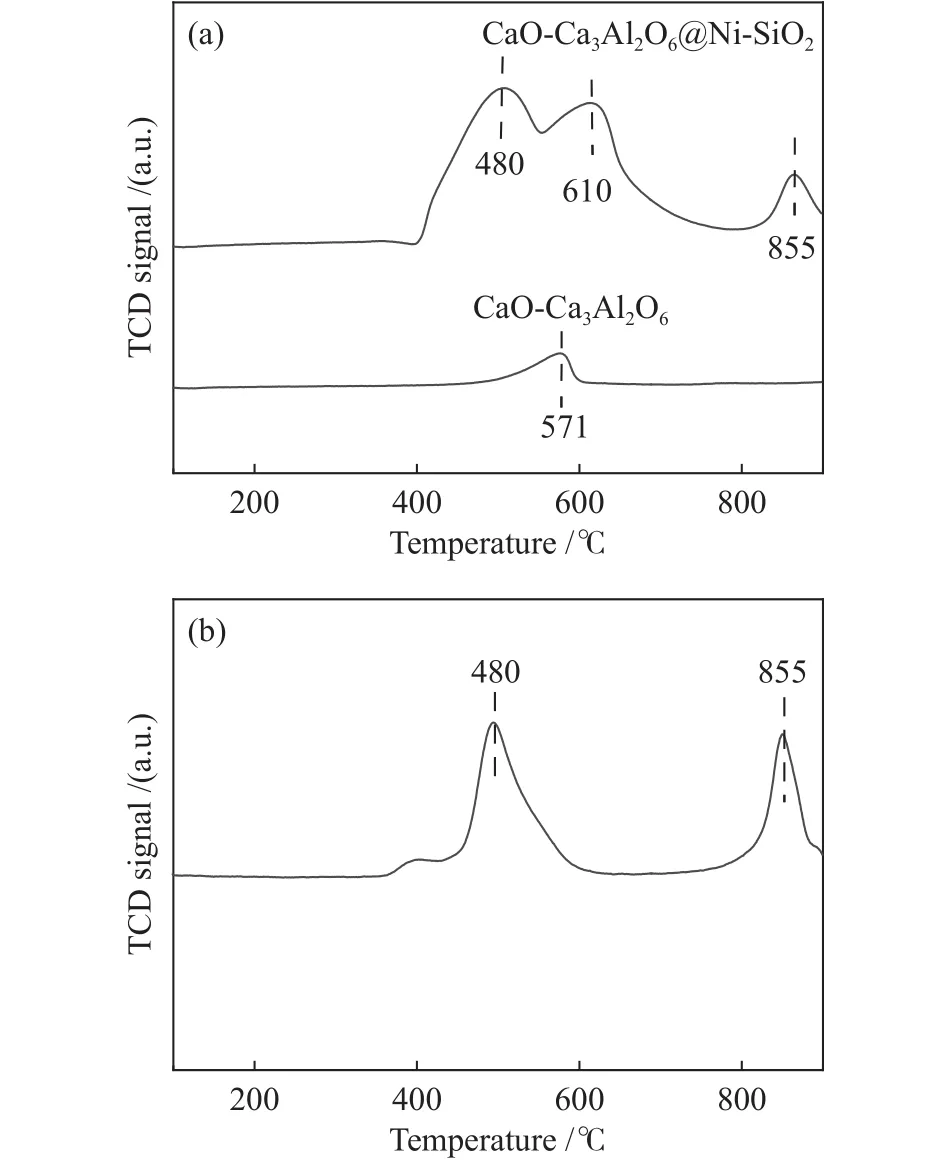
图6 不同预处理温度下复合催化剂的H2-TPR谱图Figure 6 H2-TPR profiles of composite catalysts at different pretreatment temperatures
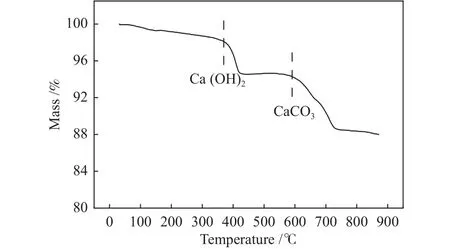
图7 复合催化剂CaO-Ca3Al2O6@Ni-SiO2的失重Figure 7 Weight loss of composite catalyst CaOCa3Al2O6@Ni-SiO2
2.3.4 形貌分析
图8是反应前后复合催化剂CaO-Ca3Al2O6@Ni-SiO2的SEM照片,从图8中可以看出,复合催化剂的主体结构呈珊瑚状,这一部分即为CaO-Ca3Al2O6@SiO2吸附组分,且从EDS能谱中可知,Ca与Al之间的分布较为紧密,说明Ca3Al2O6作为惰性组分保护CaO在吸附和脱附过程中不会发生烧结,而SiO2均匀分布在两者表面,作为一层保护壳层,使得吸附组分在反应时的体积膨胀和收缩过程都在壳层内部进行,不会造成催化剂结构上的坍塌,这一点从反应前后复合催化剂的形貌结构基本没有发生变化也可以得到充分的证明。吸附组分表面有许多乳白色的小球,这一部分是Ni@SiO2催化组分,SiO2包覆Ni,保证了Ni活性组分不会在随着催化组分再生的过程中团聚,同时也能观察到Ni在复合催化剂表面的分布非常均匀,说明还有部分Ni是负载在SiO2表面,虽然这样的结构可以提供更多的活性位点,可以使反应物与Ni活性中心更好的接触,且SiO2有效避免了吸附组分的结构变化对Ni的包埋,但这也同样导致了在多次循环过程中这一部分Ni发生了团聚,造成了复合催化剂制氢性能的下降。

图8 反应前后CaO-Ca3Al2O6@Ni-SiO2的SEM图和EDS图Figure 8 SEM and EDS mapping of CaO-Ca3Al2O6@Ni-SiO2
为进一步了解复合催化剂的形貌结构,更加直观地看到复合催化剂各组分的分布,对反应前后催化剂进行了TEM 测试,结果如图9所示。由图9可以看到,复合催化剂具有一定的包覆结构,惰性组分Ca3Al2O6起到了骨架作用,SiO2包覆在外面,同时Ni@SiO2分布在SiO2壳的外表面,可以有效防止Ni活性组分被吸附组分包埋。这种结构极大地提高了复合催化剂的循环稳定性,其中,SiO2起到了结构稳定的作用,有效解决了复合催化剂循环制氢过程中的结构坍塌问题,同时由反应后复合催化剂的TEM照片可知,10次反应后深色部分明显增加,这是因为制备的复合催化剂存在第二种形式的Ni,这一部分Ni负载在SiO2表面,没有被SiO2包覆,在循环过程中发生了团聚。

图9 反应前后复合催化剂的 TEM 照片((a)、(b)) 反应前 CaO-Ca3Al2O6@Ni-SiO2;((c)、(d)) 反应后 CaO-Ca3Al2O6@Ni-SiO2)Figure 9 TEM images of composite catalysts
综上所述可以推断出复合催化剂的结构,其结构示意图如图10所示。复合催化剂由Ni、CaO、Ca3Al2O6、SiO2四部分组成,吸附组分在内部,催化组分在外部,其中,吸附组分是以CaO-Ca3Al2O6为核,SiO2为壳的核壳结构;催化组分一部分是以Ni为核,SiO2为壳的核壳结构;另一部分Ni直接负载在SiO2壳层上。在进行SESMR制氢反应时,CH4和H2O先在最外部的催化组分上发生重整反应生成H2和CO2,然后CO2穿过SiO2壳层进入到内部被CaO-Ca3Al2O6吸附,待吸附组分吸附饱和后,将反应器温度升至750 ℃使催化剂再生,再生后的催化剂将再次在CH4和H2O气氛下进行反应。相比于直接浸渍法制备的复合催化剂,CaOCa3Al2O6@Ni-SiO2复合催化剂具有两大优势:第一,吸附组分被SiO2限制在足够大的空腔内,不会因为反应过程中的体积变化造成催化剂结构的坍塌;第二,有效防止了催化组分随着吸附组分高温再生时的团聚失活。但该复合催化剂仍有一部分Ni没有被SiO2包覆,而是暴露在壳层外表面,这一部分Ni虽然可以更好地与反应物接触,催化重整反应,但是随着吸附组分高温再生时会发生团聚,造成复合催化剂制氢性能的下降,这是下一步复合催化剂结构优化的重点。
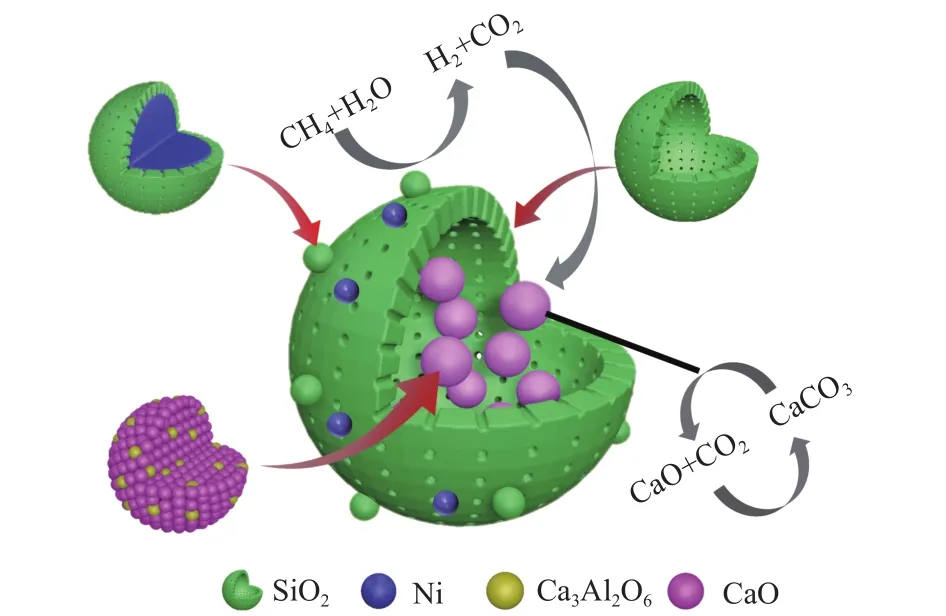
图10 CaO-Ca3Al2O6@Ni-SiO2复合催化剂的结构Figure 10 Structure of CaO-Ca3Al2O6@Ni-SiO2 composite catalyst
3 结 论
采用自模板法结合选择性刻蚀法制备出了复合催化剂CaO-Ca3Al2O6@Ni-SiO2,该复合催化剂中惰性组分含量仅为19.98%,在保证催化剂结构稳定的同时避免了因惰性组分的引入而导致的CO2吸附容量的下降,同时,SiO2的包覆使得吸附组分在吸脱附CO2的过程中的体积变化不会造成复合催化剂结构的坍塌,提高了复合催化剂的循环稳定性。
在反应温度为600 ℃,再生温度为750 ℃,压力0.1 MPa,H2O/CH4物质的量比4.8,空速968 h-1的条件下,复合催化剂制氢含量可达99.6%,10次反应后,H2的含量仍然保持在97.3%,这是因为SiO2包覆活性组分Ni防止了其在脱炭再生过程中团聚失活;但CH4转化率在10次反应内由99.5%下降至91.8%,这是因为复合催化剂的催化组分中仅有一部分是以Ni为核、SiO2为壳的核壳结构,还存在部分Ni直接负载在壳层SiO2上。
猜你喜欢
核科学与工程(2021年4期)2022-01-12 06:29:16
原子与分子物理学报(2021年2期)2021-03-29 07:30:58
新世纪智能(数学备考)(2020年9期)2021-01-04 00:25:02
家庭医学(下半月)(2020年7期)2020-08-24 07:47:02
七彩语文·写字与书法(2018年8期)2018-09-28 05:25:12
西安工程大学学报(2016年6期)2017-01-15 14:08:22
当代化工研究(2016年5期)2016-03-20 16:21:32
湖北师范大学学报(自然科学版)(2015年1期)2016-01-10 08:41:29
电源技术(2015年11期)2015-08-22 08:50:26
低温与特气(2014年4期)2014-03-20 13:36:50
