
11 例原发性肾性糖尿患儿SLC5A2 基因变异分析
2022-12-28 08:28:40万灵陈朝英涂娟李华荣
临床儿科杂志 2022年12期
万灵 陈朝英 涂娟 李华荣
首都儿科研究所附属儿童医院(北京 100020)
原发性肾性糖尿(primary renal glucosuria,PRG)是由于近段肾小管对葡萄糖的重吸收障碍引起的糖尿,临床特点是血糖浓度正常,不伴有肾小管的其他功能异常,发病率为1/20000[1]。其又称为家族性肾性糖尿(familial renal glucosuria,FRG),是一种常染色体不完全外显的共显性遗传病。绝大多数的PRG患者与Na+-葡萄糖协同转运蛋白2(sodium-glucose co-transporter 2,SGLT 2)的编码基因SLC 5 A 2变异有关(OMIM 182381)[2-4]。为研究PRG 患儿SLC 5 A 2基因变异情况及基因型与表型的关系,本文对11例临床符合肾性糖尿的患儿进行SLC5A2基因分析。
1 对象及方法
1.1 研究对象
收集2013—2021年确诊于首都儿科研究所附属儿童医院的PRG患儿。纳入标准:①发病年龄0~16岁;②临床符合PRG诊断标准[2,5];③有基因检测结果。
本研究经首都儿科研究所附属儿童医院伦理委员会批准。
1.2 方法
1.2.1 资料收集 根据病历资料收集PRG患儿的人口学信息、临床资料及基因检测结果。
1.2.2 诊断标准 PRG诊断参照文献[2,5]:①24 h尿葡萄糖>0.3 g/1.73 m2;②葡萄糖代谢正常;③无其他肾脏病证据,血尿、蛋白尿、氨基酸尿或磷酸盐尿等,肾功能正常。
糖尿定义:24h尿糖≥0.3 g/1.73m2,<10 g/1.73m2定义为轻度糖尿,≥10 g/1.73m2为重度糖尿。但由于小婴幼儿留取24 h尿标本困难,故本研究将3次以上晨尿常规发现尿葡萄糖+~++者定义为轻度糖尿,尿葡萄糖+++~++++者定义为重度糖尿。
1.2.3 基因检测 取患儿及其父母的外周静脉血,提取基因组DNA,送金准基因公司进行高通量测序。测序结果与人类基因变异数据库(Human Gene Mutaion Database,HGMD)、千人基因组数据库、gnomAD 数据库、本地人频率数据库对比,将在HGMD 数据库中未查到,且千人基因组变异频率<5%、本地人群频率<2%的基因变异确定为新发现的变异。变异蛋白根据ACMG 指南进行变异蛋白的致病性预测,采用在线软件SIFT、PolyPhen2和Mutation Taster 评估和分析错义变异的致病性,Clustal X软件进行同源序列比较,以区分保守序列和非保守序列,Human Splicing Finder软件预测剪接变异意义,PyMOL软件(http://www.pymol.org)生成SGLT2蛋白野生型及变异型的三维结构图。
2 结果
2.1 一般情况
符合纳入标准共11例患儿,其中男5例,女6例,男/女比例为1:1.2。诊断年龄为生后1 天~13 岁,中位年龄29 月龄,随访病程时长中位数为40 个月(5~105个月)。
11例患儿均无糖尿病家族史,5例有尿糖阳性家族史。2例因尿频、2例因尿色浑浊黏稠起病就诊,其余均为体检或因其他疾病住院意外发现尿糖阳性。轻度糖尿2例,重度糖尿9例,均无多饮、多尿、消瘦、出汗、心悸等表现,生长发育无异常。除例8、例11尿钙略高[分别为0.11mmol/(kg·d)、0.1mmol/(kg·d)]外,所有患儿血生化、糖化血红蛋白、空腹及餐后2 h血糖、胰岛素、C肽、尿电解质、泌尿系超声等结果均正常,其他基线信息见表1。
2.2 SLC5A2基因测序结果
11 例患儿未发现纯合变异,杂合变异比例为36%(4/11例),复合杂合变异比例为64%(7/11例),共发现14种变异,其中有10种错义变异、1种缺失变异、3种剪接变异(表1)。ACMG指南进行变异蛋白的致病性预测,结果均为VVS(临床意义未明)。见表2。
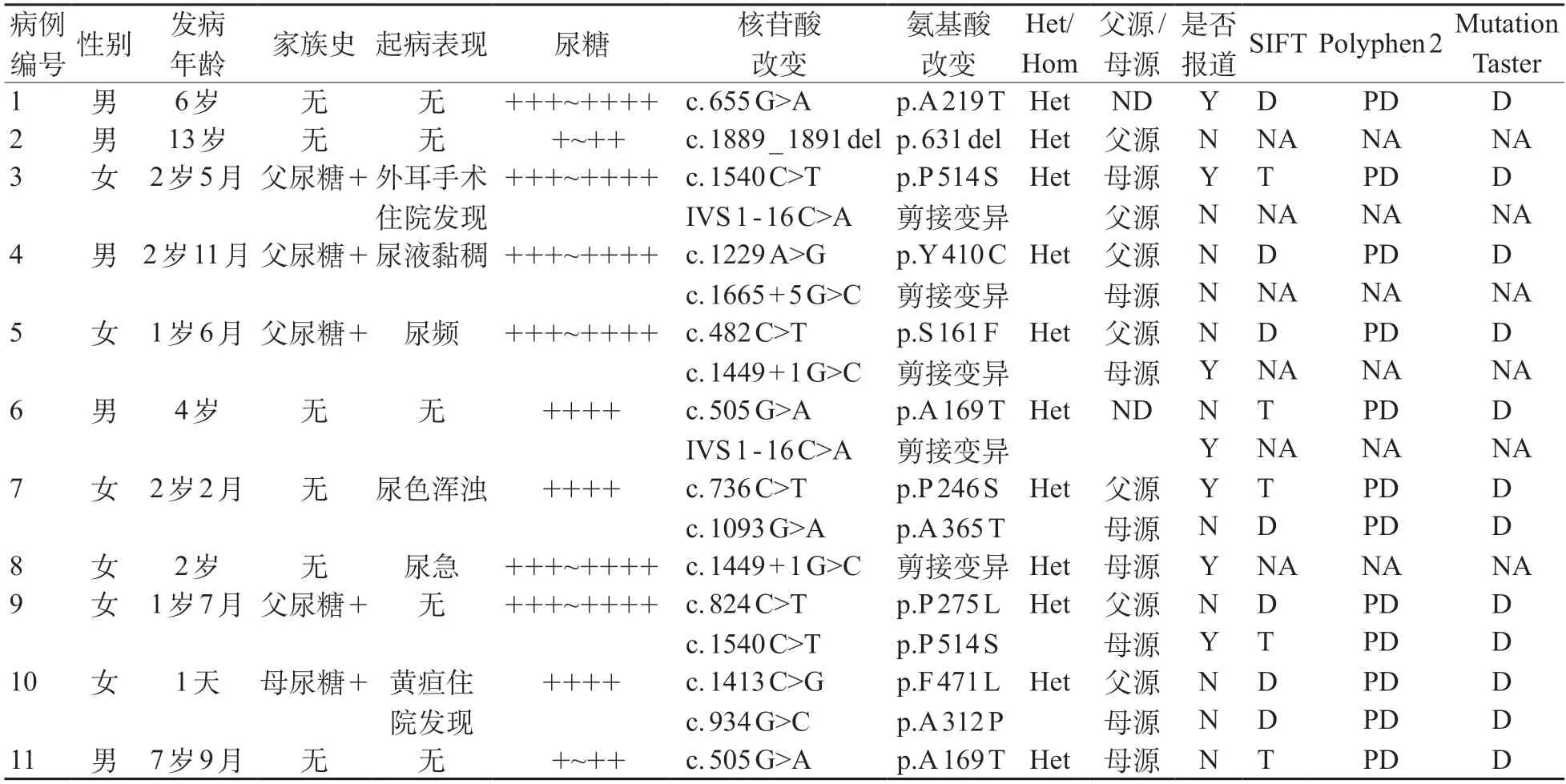
表1 11例PRG患儿临床资料及SLC5A2基因变异分析

表2 ACMG指南进行变异蛋白的致病性预测
错义变异中,c.655G>A(p.A219T)、c.736C>T(p.P246S)、c.1540C>T(p.P514S)为前期文献已报道的致病变异,其余c.482C>T(p.S161F)、c.505G>A(p.A 169 T)、c.824 C>T(p.P 275 L)、c.934 G>C(p.A 312 P)、c.1093 G>A(p.A 365 T)、c.1229 A>G(p.Y 410 C)、c.1413 C>G(p.F 471 L)7 种均为新发现变异。3 种在线软件SIFT、PolyPhen 2和Mutation Taster预测p.S161F、p.P275L、p.A312P、p.A365T、p.Y410C、p.F471L 6种变异均存在高度致病可能;PolyPhen2和Mutation Taster预测p.A169T为致病性变异,但SIFT软件预测为良性变异。用Clustal X软件对所有错义变异位点进行不同物种同源序列分析,均显示为高度保守序列(图1)。用PyMOL软件生成SGLT2蛋白野生型及变异型的三维结构图(图2),发现新发现的7种错义变异均可破坏SGLT2蛋白的局部二级结构、影响蛋白的正常功能。
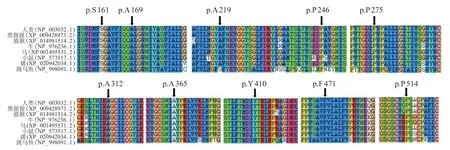
图1 10 种SGLT2 错义变异在8 种生物中的同源保守序列分析图
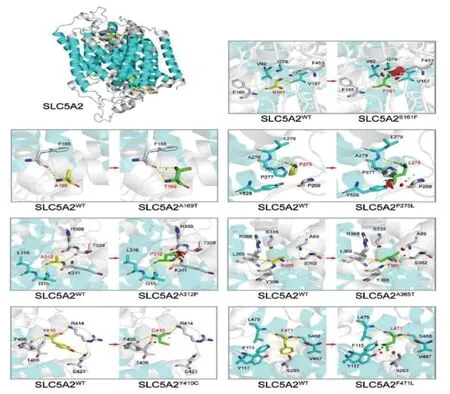
图2 SGLT2 蛋白野生型及7 种新发现的错义变异型的三维结构图(WT:野生型)
缺失变异c.1889_1891del为新发现的变异,该变异导致第631号氨基酸缺失(p.631del)。
剪接变异为IVS 1-16 C>A、c.1665+5 G>C、c.1449+1G>C。其中c.1665+5G>C为新发现的变异,生物信息分析软件Human Splicing Finder预测可能会影响mRNA的正常剪接,该变异临床意义未明;IVS1-16C>A、c.1449+1G>C为前期文献报道变异,为致病性变异。
3 讨论
肾性糖尿是由于血浆葡萄糖浓度超过肾糖阈值,导致尿中葡萄糖浓度增高所致的临床综合征。肾脏对葡萄糖代谢的调节作用主要包括糖异生、肾小球对葡萄糖滤过和近曲小管对葡萄糖重吸收。因此尿液中葡萄糖应为阴性。当血浆葡萄糖浓度达到近10 mmol/L时,将超过肾糖阈值,导致尿中可检测到葡萄糖[6]。研究表明,主要有两种钠-葡萄糖协同转运蛋白,即SGLT1和SGLT2,参与肾脏对葡萄糖的重吸收[7]。SGLT2特异性地表达于肾脏近曲小管的S 1 段,具有低亲和力、高容量的特点,是肾脏近曲小管重吸收葡萄糖的关键,80%~90%的葡萄糖由SGLT2转运蛋白重吸收,钠离子与葡萄糖的偶联比例为1:1[8]。SGLT1主要表达于肠黏膜上皮细胞的刷状缘,在肾脏近髓质的近端小管S 3 段也有表达,具有高亲和力、低容量的特点,未被SGLT 2 转运的10%~20%的葡萄糖到达S3段被SGLT1重吸收,钠离子与葡萄糖的偶联比例为1:2。SGLT 1 和SGLT 2二者协同合作以完成原尿中葡萄糖的重吸收[2,4,9]。
2000 年,Santer 等[10]首次报道在PRG 患者中发现SGLT2蛋白的编码基因SLC5A2变异,此后一系列研究证实SLC 5 A 2变异是导致FRG 的主要原因[3-4,7-8,11]。SLC5A2基因位于染色体16p11.2,全长7.7kb,包含14个外显子,编码672个氨基酸组成的肽链[12]。目前国内外报道的SLC5A2基因变异有90多种(人类基因变异数据库,HGMD professional 2021),包括错义变异、无义变异、剪接变异、小片段缺失/插入变异等,其中IVS 7+5G>A是热点变异[13]。邵乐平课题组对7个中国FRG家系进行SLC5A2基因检测,发现其中5个家系共11例患者均存在c.886(-10--31)del变异,等位基因变异频率高达43%,据此推测SLC5A2基因c.886(-10--31)del变异可能是中国人群的高频变异[14,15]。Yu等[8]对10个FRG家系研究,发现IVS1-16C>A和c.886(-10--31)del可能是高频剪接位点变异,当在SLC5A2外显子中找不到变异时可以优先筛选。Dorum 等[11]对16例FRG 患者进行SLC 5 A 2基因检测,发现最常见的变异为c.655G>A。
本研究发现的10种错义变异中,p.A219T[16-17]、p.P246S[8,18]、p.P514S[8,18-21]为前期文献报道的已知致病变异,p.S161F、p.A169T、p.P275L、p.A312P、p.A365T、p.Y410C、p.F471L共7种变异为新发现的变异。ACMG指南预测其变异临床意义未明,但3种在线软件SIFT、PolyPhen2和Mutation Taster对变异位点进行致病性预测,除SIFT软件预测p.A169T为良性变异,其余均为致病性变异或可能致病性变异,ClustalX软件进行不同物种同源序列分析,显示所有错义变异位点均为高度保守序列。利用PyMOL软件生成SGLT 2 蛋白野生型及变异型的三维结构图发现:①在野生型蛋白中,161位丝氨酸与165位苯丙氨酸、157 位缬氨酸之间存在极性相互作用,p.S 161 F 破坏了与缬氨酸之间的极性相互作用,并对周围的G79、V82、V157及F453之间产生巨大的斥力;②p.A 169 T 可引起第169 位丙氨酸变异为苏氨酸,破坏了其与165位苯丙氨酸的极性相互作用;③p.P275L可引起第275位脯氨酸变异为亮氨酸,可对周围的P269、P277、Y528产生巨大斥力;④在野生型蛋白中,A312与T308、I315及L316之间存在极性作用,p.A312P可导致312位丙氨酸变异为脯氨酸,破坏与T308之间的极性,且P312与H309之间可产生一个新的相互作用,对周围的T 308、H 309、K311之间产生巨大的斥力、破坏氨基酸的二级结构;⑤在野生型蛋白中,A 365 与S 336、Y 366、R 368 及L 369 之间存在极性相互作用,p.A 365 T 可引起365位丙氨酸变异为苏氨酸,T 365 与Y 366 之间产生了新的极性相互作用,且T365与A89、S362之间产生斥力、破坏这些氨基酸所在的二级结构;⑥野生型蛋白中,Y410与F405、T406、R414及E421之间存在极性作用,p.Y410C可破坏与E421之间的极性作用,且变异后的C 410与T406 之间可产生排斥力、破坏二级结构;⑦p.F471L引起第471位苯丙氨酸变异为亮氨酸,对周围的F113、Y117、S293及L475产生斥力,破坏了这些氨基酸所在的二级结构。故推测新发现的7种错义变异均有致病性。缺失变异c.1889_1891del为新发变异,导致第631 号氨基酸缺失。剪接变异IVS1-16C>A[8,22]、c.1449+1G>C[18,22]为前期文献报道的已知致病变异,c.1665+5G>C为新发变异,生物信息分析软件Human Splicing Finder预测可能会影响mRNA的正常剪接。
多项研究表明,在PRG患者中,无论何种变异(如错义变异、无义变异或剪接位点变异),杂合变异个体多表现为轻度糖尿,而纯合变异或复合杂合变异患者通常表现为重度糖尿[23-24]。本研究中例1、2、8、11以及例3、4、5、9的父亲和例10的母亲均为杂合变异,其中仅例1、8表现为重度糖尿,其余均为轻度糖尿,而例3、4、5、6、7、9、10均携带复合杂合变异,都表现为重度糖尿,与前期文献报道相符。这可能是由于杂合变异导致单倍剂量不足,肾小管上皮细胞正常表达的SGLT2数量减少,导致葡萄糖重吸收减少而呈现少量糖尿;而复合杂合变异可导致更严重的近端小管的重吸收障碍。
本研究中除例1的父母外,其余父母均进行基因检测。例2、8、11基因变异均来源于父母其中一方,但携带相同变异的父亲或母亲多次尿检葡萄糖均为阴性,说明SLC5A2相同的基因型可以有不同的表型。携带复合杂合变异的例3、4、5、6、7、9、10,其携带的2个变异位点均分别来源于父母,但仅例3、4、5、9的父亲和例10的母亲有轻度糖尿表现,其余携带杂合变异的父母多次尿检均无尿糖,符合既往学者提出的SLC5A2基因存在不完全外显的共显性特征[24-25]。
此外,例1、11虽然都为杂合错义变异,但例1表现为重度糖尿,例11为轻度糖尿。分析发现,例1携带的变异c.655G>A 为已报道的常见致病变异[11],可引起第219 位氨基酸由丙氨酸变异为苏氨酸,SIFT、PolyPhen2和Mutation Taster等3种软件预测致病性均考虑高度致病。而例11携带的变异c.505G>A为新发现的变异,仅PolyPhen2和Mutation Taster软件预测为致病性变异,SIFT软件预测为良性变异,且蛋白质的三维结构图显示,变异引起第169位丙氨酸变异为苏氨酸,仅可破坏与165位苯丙氨酸的极性作用,破坏范围较小。由此说明,虽然变异类型相同,但不同位点的变异,表型可有明显差异。
例2 为缺失变异,临床表现为轻度糖尿,而其携带相同变异的父亲则临床无尿糖表现,分析发现c.1889_1891 del 可导致第631 号氨基酸缺失(p.631del),该位点位于氨基酸序列的末端,考虑可能对编码的SGLT2蛋白功能影响较小,或者其正常的等位基因可代偿一部分葡萄糖重吸收功能,故患儿与其父亲表型不同。
本研究共发现了2 例患儿携带了文献报道的高频剪接位点变异IVS 1-16 C>A,1 例患儿携带c.655G>A,其余绝大多数变异仍然是某个PRG家族特有和专属的,与既往研究一致[2,8,11],未发现变异热点IVS 7+5G>A和c.886(-10--31)del,考虑可能与本研究的病例数较少有关,需要后期扩大样本量分析。前期国内外的研究大多数均基于成人PRG,发现其预后较好,一般无需特殊治疗,只需适当增加碳水化合物的摄入量,而关于儿童期发病的PRG的研究极少。本研究对所有患儿进行了随访,最长随访时间为105个月,临床均无糖尿病“三多一少”症状或全身乏力表现,生长发育正常,无严重并发症。但也有文献报道,PRG患者在妊娠期间出现尿糖增多,可出现渗透性利尿而导致多尿、脱水甚至意识丧失[26],在极度饥饿时可出现酮症[27],因此仍需定期随访。
猜你喜欢
广东药科大学学报(2023年5期)2023-12-30 00:08:39
时代报告·奔流(2022年1期)2022-04-29 04:10:56
昆明医科大学学报(2022年3期)2022-04-19 13:59:42
成都医学院学报(2021年2期)2021-07-19 08:35:32
种子(2021年3期)2021-04-12 01:42:22
华声文萃(2019年8期)2019-09-10 07:22:44
文萃报·周二版(2019年25期)2019-09-10 07:22:44
外语教学理论与实践(2016年1期)2016-06-11 05:51:48
浙江医学(2014年17期)2014-04-13 10:13:16
华东理工大学学报(自然科学版)(2014年1期)2014-02-27 13:48:29
