
蛋白质二硫键异构酶a3的表达纯化、活性测定与小分子抑制剂的筛选
2022-12-25 07:30李海涛周鹤峰
遵义医科大学学报 2022年6期
宋 煜,李海涛,邵 敏,周鹤峰
(1.遵义医科大学珠海校区 生物工程教研室,广东 珠海 519000;2.遵义医科大学珠海校区 生物化学教研室,广东 珠海 519000)
二硫键即-S-S-(Disulfide bond,Dsb),由多肽链上不同位点的两分子巯基(-SH)氧化形成,可分为链内和链间二硫键[1]。在生理条件下,蛋白质的二硫键经蛋白质二硫键异构酶催化作用断裂形成两个巯基,其功能发生变化[2]。在真核生物中,二硫键通常是在粗面内质网而非原生质形成[3]。二硫键能够被还原剂裂解是其最重要的一个特性。能够将二硫键裂解的还原剂较多,其中硫醇如β-硫基乙醇或二硫苏糖醇(Dithiothreitol,DTT)较为常用。在体内和人工条件下,蛋白质二硫键异构酶 (Protein disulfide isomerase,PDI)能够催化巯基与二硫键之间的转化,具有分子伴侣功能[4]。二硫键与巯基两种基团之间的相互转化主要由巯基-二硫键氧化还原酶作为反应的催化剂[5],是一类硫醇氧化还原酶,作为内质网中的一种驻留蛋白[6],它催化新生多肽中二硫键的形成和异构化,这一过程对于正确的蛋白质折叠和维持细胞内蛋白质稳态至关重要[7-8]。蛋白质二硫键异构酶a3(PDIA3),又被称作ERp57,分子量为58 KDa,具有PDI酶活性,可通过促进蛋白底物中二硫键的形成来调节新合成蛋白质的折叠[9]。
近年来,PDI因其参与多种生理和病理过程而成为治疗多种疾病和癌症的新靶点[10-11],例如,最近的研究表明PDI家族在调节血栓形成中起重要作用,促进血小板聚集和血栓形成[12]。鉴于PDI在血栓形成中扮演的重要角色,调节PDI活性可以作为治疗血栓的潜在药物靶点。抑制血小板的聚集和纤维蛋白的生成可通过抑制PDI活性来实现[13-14]。PDIA3是PDI家族的主要成员之一,与PDI具有高度同源性[15]。由于PDIA3可以调节多种刺激物诱导的血小板聚集和血栓形成,因而可研究PDIA3抑制剂来抑制血栓形成[16]。
由于PDIA3具有断裂二硫键使其还原为巯基的能力,而牛胰岛素分子的A链与B链之间通过两对二硫键连结[17],本实验采用牛胰岛素浑浊法测定PDIA3的活性。在还原剂DTT存在条件下,PDIA3将牛胰岛素A链和B链之间的二硫键还原为巯基。A、B链之间的二硫键还原并断裂后,A、B链发生错误折叠,B链形成沉淀物,通过测定吸光度即可知其还原活性。
虚拟筛选(Virtual screening,VS)是以药物设计理论为基础,借助计算机技术和专业应用软件,从大量化合物中筛选出一些有前途的化合物,进行评估实验活性的方法[18]。虚拟筛选作为计算机辅助药物研究设计中的一项实用化技术,在当代的创新药物研发中发挥着重要作用[19-20]。因此,本研究拟提取人总RNA,利用mRNA逆转录合成cDNA,并以此为模板克隆PDIA3基因的编码区序列,进而诱导表达并纯化出PDIA3,从Specs商业化合物库中以PDIA3为靶点筛选小分子抑制剂,并对筛选到的化合物进行抑酶活性测定,为PDI蛋白抑制剂的筛选和抗血栓药物的研发奠定基础。
1 材料与方法
1.1 材料 pET28a(+)来自Addgene质粒库,Escherichia coli(E.coli) DH5α,E.coliBL21(DE3),HEK293细胞均来自生物工程教研室贮存。T4 DNA连接酶,Taq DNA聚合酶,BamH I核酸内切酶和NotI核酸内切酶均购自New England Biolabs公司;DL2000 DNA Marker和Protein SDS-PAGE loading buffer,逆转录试剂盒RR055A购自Takara生物公司;彩虹245广谱蛋白Marker,DTT,牛胰岛素,BCA试剂盒和SDS-PAGE 凝胶试剂盒购自Solarbio公司;异丙基-β-D-硫代半乳糖苷(IPTG),考马斯亮蓝R-250及DNA上样缓冲液购自宝生物工程(大连)有限公司。
PCR仪,全自动凝胶成像分析系统及高电流型电泳电源购自伯乐生命医学产品有限公司;全波段酶标仪购自美国ThermoScientific公司;核酸、蛋白电泳仪购自北京六一生物科技有限公司;低温超声破碎仪购自上海兰仪实业有限公司;镍离子柱-FPLC(1 mL)购自Takara生物公司;超净工作台,电热恒温培养箱,恒温摇床和电热恒温水浴锅购自上海博迅实业有限公司;低温冷冻离心机购自德国Eppendorf公司;掌上离心机购自Solarbio公司。
1.2 方法
1.2.1 HEK293细胞总RNA的提取 将含HEK293细胞的培养皿置于冰上,用冷PBS洗涤细胞两次,加入1 mL Trizol裂解液裂解细胞,将溶液移至1.5 mL Ep管,室温静置5 min。然后加入0.2 mL氯仿涡旋15 s,室温静置2 min后,4 ℃、12 000 g离心15 min。取上清加入0.5 mL异丙醇,涡旋混合,室温培养5 min。4 ℃、10 000 rpm离心10 min,去除上清,加1 mL 75%乙醇洗涤沉淀,4 ℃、14 000 r/min离心5 min,弃上清,待沉淀的RNA在室温环境下自然干燥后,加50 μL DEPC水溶解RNA沉淀。然后取样10 μL进行琼脂糖凝胶电泳鉴定,剩余样品-20 ℃保存用于逆转录合成cDNA基因。
1.2.2 pET28a-PDIA3载体构建及鉴定 基于GenBank中PDIA3的基因序列,使用Vector-NTI软件设计引物,由广州艾基生物科技有限公司完成引物的合成。RT-PCR法扩增目的基因PDIA3的编码区序列,用限制性内切酶BamH I和NotI进行酶切,得到具有互补粘性末端的目的基因和载体,通过T4 DNA连接酶的催化作用进行连接,然后转入E.coliDH5α中,涂布在具有卡那霉素(Kana)抗性的固体LB培养基上。挑取阳性单克隆菌落在具有Kana抗性的液体LB培养基中扩大培养,提取质粒送公司测序,经测序确定重组质粒pET28a-PDIA3构建成功后,转入E.coliBL21(DE3)中,得到含有重组质粒pET28a-PDIA3的工程菌。
1.2.3 重组蛋白的诱导表达 重组蛋白PDIA3采用大肠杆菌表达系统和纯化[21]。将含重组质粒pET28a-PDIA3的工程菌置于含Kana 100 μg/mL的5 mL LB培养基中振摇培养至OD600nm约为0.5(37 ℃,220 r/min),取出1.5 mL至Ep管作为诱导前样品,然后用IPTG(1.0 mmol/L)分别诱导2 h(取出1.3 mL)、4 h(取出1 mL)、6 h(取出1 mL),同时将空载体pET28a于同样条件下培养6 h后取出1 mL作为对照。每次取样后,4 ℃、12 000 r/min离心1 min,弃上清,100 μL 1×PBS洗涤菌体一次,100 μL 1×PBS悬浮,再加等体积2× Protein SDS-PAGE loading buffer混匀,立即煮沸(100 ℃、1 min),然后进行SDS-PAGE电泳。
1.2.4 工程菌的扩大培养 将活化后的种子液按10%比例接入200 mL Kana LB液体培养基(剩余种子液取出1 mL作为阴性对照),振摇培养至OD600nm约为0.5(37 ℃,220 r/min),加入IPTG(1.0 mmol/L)诱导4 h(取出1 mL作为阳性对照),收集菌体,4 ℃、4 000 r/min离心10 min,弃上清,加入30 mL 1×PBS洗涤菌体一次,再4 ℃、4 000 r/min离心10 min,弃上清,加20 mL Binding buffer悬浮菌体,用于超声破碎。
1.2.5 超声破碎 超声波破碎上述悬浮菌体(工作2 s,间隔2 s,功率40%,20 min),破碎后的菌体4 ℃、10 000 r/min离心30 min。离心后上清用于纯化,沉淀用作对照。
1.2.6 镍柱亲和层析纯化 装柱:吸300 μL混匀的50% NI-NTA至1 mL层析柱中,待完全沉淀后,加入2 mL去离子水洗涤一次(流速:重力作用);镍柱平衡:加入2 mL Binding buffer进行平衡;蛋白上样:将样品重复上柱(5次);洗脱:①杂蛋白用5 mL含20 mM咪唑的Washing buffer洗脱;②目的蛋白用0.5 mL 含250 mM咪唑的Elution buffer洗脱。
1.2.7 PDIA3蛋白的生物学活性测定 比活力是在特定条件下,每毫克蛋白质或RNA所具有的酶活力单位数[22]。纯化的重组蛋白PDIA3用牛胰岛素浑浊法测定其生物学活性[23]。在96孔板中,对照组每孔加入10 μL缓冲液,实验组每孔加入10 μL 0.04 mg/mL PDIA3[24],然后每孔分别加入50 μL 1 mg/mL胰岛素、10 μL 1.5 mM DTT,终体系为70 μL,每组实验设计3个复孔。用酶标仪分别于0、20、40 min 630 nm处测定混合体系的OD值。PDIA3蛋白酶比活力单位采用Ibbetson[25]定义。
1.2.8 PDIA3小分子抑制剂的筛选与体外抑酶活性测定 从PDB数据库中下载PDIA3的X-射线3D结构(PDBID:3F8U),去除PDIA3结构中多余的水分子,加入氢离子,将蛋白结构能量最小化,选择PDIA3第二活性位点(C406-G407-H408-C409)作为分子对接中的活性位点,选择Specs商业化合物库通过MOE软件处理进行分子对接,计算 PDIA3与小分子化合物之间的结合能,进行筛选。
对筛选到的化合物进行体外抑酶活性测定。在 96 孔板中,对照组每孔加入10 μL 0.04 mg/mL PDIA3和10 μL缓冲液,实验组每孔加入10 μL 0.04 mg/mL PDIA3和10 μL不同浓度的待测化合物,待测化合物浓度分别为1、3、10、30和100 μM,然后每孔分别加入50 μL1 mg/mL胰岛素、10 μL 1.5 mM DTT,终体系为80 μL,每组实验设计3个复孔。25℃孵育84 min用酶标仪630 nm处测定混合体系的OD值。抑制PDIA3酶活性的计算公式为[26]:酶活性抑制率(%)=[1-(OD[化合物+PDIA3+DTT]-OD[DTT])/(OD[PDIA3+DTT]-OD[DTT])]×100%。
2 结果
2.1 HEK293细胞总RNA的提取 Trizol法提取HEK293细胞的总RNA,通过琼脂糖凝胶电泳鉴定其完整性。图1显示,两条泳道均为RNA样品,28S rRNA、18S rRNA、5S rRNA 3条带自上而下分布,其中28S rRNA条带的亮度约为18S rRNA条带的两倍,5S rRNA条带相对模糊,表明RNA提取较完整。
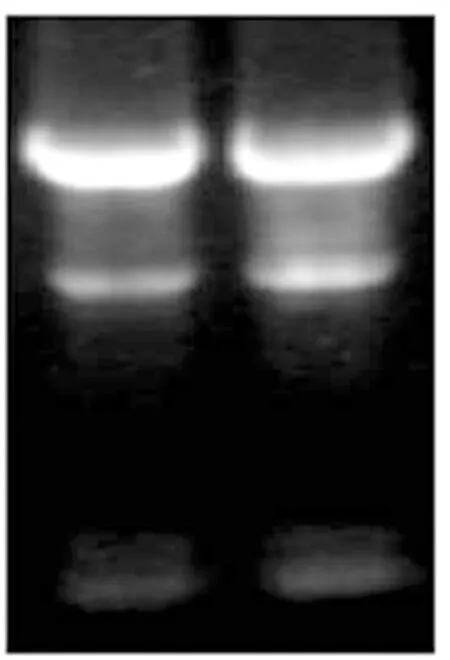
图1 RNA琼脂糖凝胶电泳鉴定结果
2.2 pET28a-PDIA3载体构建及鉴定 广州艾基生物科技有限公司完成引物的合成,上游引物5’-TTCGGGATCCTCCGACGTGCTAGAACTCACG-3’,下游引物5’-AGAATGCGGCCGCATTAGAGATCCTCCTGTGCCTTCTTC-5’,其中,划线部分分别为BamH I和Not I的识别序列。为了构建pET28a-PDIA3重组质粒,利用RT-PCR扩增获得了PDIA3的cDNA,其大小与理论值1518 bp相符(见图2),通过酶切、连接、转化等获得克隆菌株。对获得的克隆菌株进行质粒提取,并利用菌落进行PCR鉴定,如图3所示,7个菌落均扩增出与理论值相符的条带。随机挑取3个菌落进行测序,测序结果显示插入的PDIA3基因序列与目的基因序列完全一致,表明PDIA3基因已成功克隆到原核表达载体pET28a(+)中。
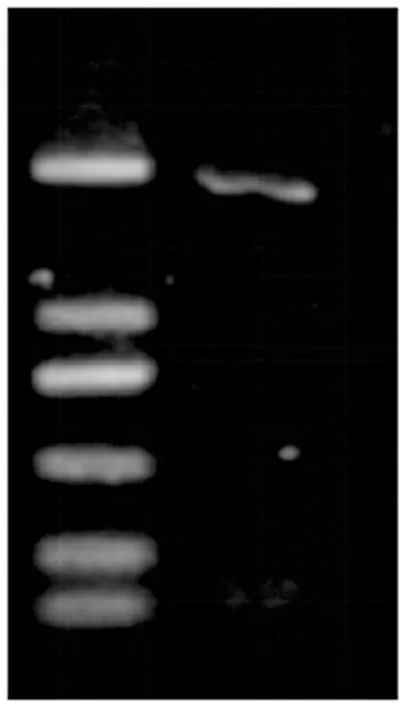
图2 RT-PCR扩增目的基因琼脂糖凝胶电泳鉴定结果
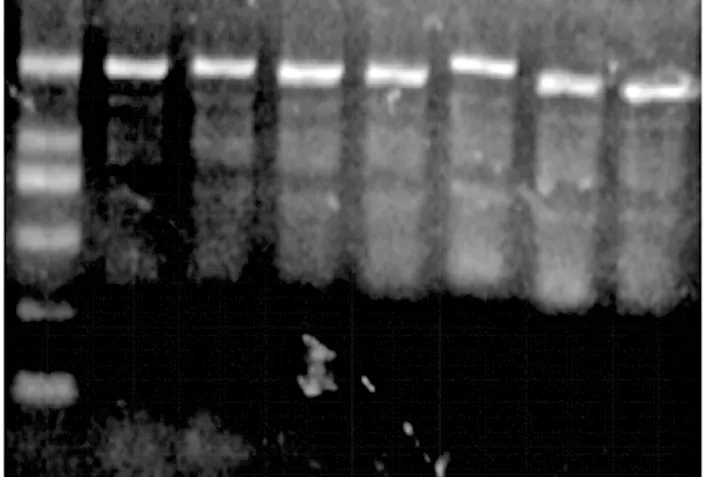
图3 菌落PCR鉴定目的基因琼脂糖凝胶电泳鉴定结果
2.3 重组蛋白的诱导表达 成功构建重组质粒后,转化BL21,挑选单克隆菌株,在含有Kana的LB培养基中培养至OD600nm约为0.5时,取出1.5 mL菌液,然后用IPTG(1.0 mmol/L)分别诱导2、4、6 h。为了作对照,同时将空载体于相同条件下培养6 h。对空载体、诱导前以及诱导2、4、6 h样品依次进行SDS-PAGE鉴定,图4的SDS-PAGE结果显示,诱导前与诱导后相比,大肠杆菌总蛋白在诱导后于48 KDa到63 KDa之间出现了新的条带,其分子量大小约为58 KDa,与预期的目的蛋白PDIA3(505个氨基酸)基本吻合。
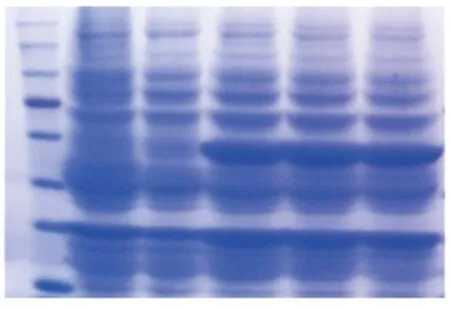
图4 重组蛋白PDIA3诱导表达后SDS-PAGE鉴定结果
2.4 重组蛋白的纯化 将经IPTG(1.0 mmol/L)诱导表达的菌体进行超声破碎,4 ℃、10 000 r/min离心30 min分别得到上清和沉淀。上清作为蛋白样品用于纯化,沉淀用作对照。由于目的蛋白含有his标签,可以通过Ni-NTA树脂亲和层析纯化。填充好镍柱后,上清液上柱,重复5次,先用5 mL 洗涤缓冲液去除杂蛋白,然后用0.5 mL 洗脱缓冲液将目的蛋白洗脱下来。对超声前后的菌体、纯化超声后上清、超声后沉淀依次进行SDS-PAGE鉴定,图5中SDS-PAGE结果显示,目的蛋白经纯化后的几乎不含杂蛋白,浓度相对较高。

图5 目的蛋白PDIA3纯化后SDS-PAGE鉴定结果
2.5 PDIA3蛋白的生物学活性测定 用牛胰岛素检测纯化蛋白PDIA3的生物学活性。从测定结果(见图6)中可以明显看出,随着反应时间的延长,实验组在630 nm处的OD值逐渐增加,表明纯化的PDIA3蛋白具有催化牛胰岛素中二硫键断裂还原成巯基的能力,BCA测定标准曲线为y=0.00125x+0.119,R2=0.999,根据OD562nm计算得到PDIA3的比活力为4.17 U/mg, PDIA3蛋白活性较好[27]。
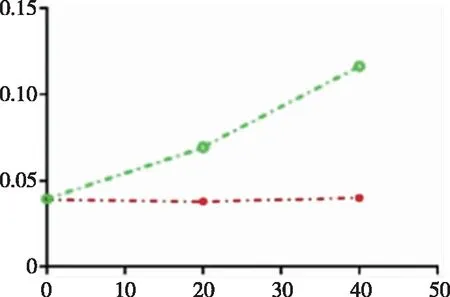
图6 PDIA3的生物学活性测定
2.6 虚拟筛选及体外抑酶活性测定结果 以PDIA3为靶点蛋白,用MOE软件将 PDIA3与库中化合物进行分子对接,从中筛选到2个对PDIA3活性有抑制作用的小分子化合物,并对筛选到的化合物用胰岛素比浊法进行PDIA3抑酶活性进行测定,图7中实验结果显示化合物去甲氧基姜黄素(Demethoxycurcumin)和 41974954对PDIA3活性有抑制作用,随着化合物浓度上升,其对PDIA3活性的抑制效果越好。化合物去甲氧基姜黄素和41974954对PDIA3活性的半数有效抑制浓度IC50分别为(6.37±0.34、129.8±26.50)μmol/L。
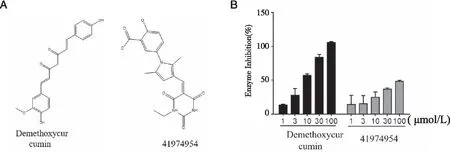
A:去甲氧基姜黄素、41974954的结构;B:两种化合物对PDZA3活性的抑制结果比较。
3 讨论
PDI是参与蛋白质折叠的关键酶,其不仅调节内质网中肽链的折叠,还与体内的血栓形成有关,是治疗血栓的潜在药物靶标[28-29]。鉴于PDI在体内血小板聚集和血栓形成中扮演的重要角色,测定PDI还原酶活性,筛选PDI抑制剂,已成为一种全新的抗血栓治疗策略[30]。
本研究基于PDIA3基因重组质粒的构建、表达纯化,测定PDIA3还原酶活性,以PDIA3为靶点,运用MOE软件对Specs商业化合物库进行虚拟筛选。其中,测定的PDI还原酶活性为4.17 U/mg,测定结果与张婷婷制备的重组PDI比活力测定结果4.3 U/mg相差不大[31],但与卢临萍[32]制备的共表达菌株PDI比活力测定结果107.2 U/mg相差较大。可作为该实验进一步完善的方向。
虚拟筛选的优点在于技术发展成熟、筛选量大且筛选成本相对低廉[33],广泛应用于基因工程与药物研发领域[34-35]。常用的化合物库有Specs商业化合物库、剑桥晶体结构数据库和世界药物索引(World drug index,WDI)等[36],本实验运用Specs商业化合物库对PDIA3小分子抑制剂进行筛选,通过虚拟筛选得到去甲氧基姜黄素和 41974954,对PDIA3活性有一定抑制作用,且化合物浓度与PDIA3活性抑制效果成正比,筛选结果与邹佳使用的Specs商业化合物库筛选结果相符[37],进一步表明此方法可以运用在大型化合物库中进行筛选。近年来,国内外在一些重要疾病的药物靶标筛选已有成功使用虚拟筛选的案例[38-39]。本研究成功筛选到蛋白PDIA3的小分子抑制剂去甲氧基姜黄素和 41974954,进一步证实PDI可作为抗血栓药物的筛选靶点,为PDI作为抗血栓治疗的潜在药物靶标的进一步发展提供了基础数据支持。
本实验通过RT-PCR技术成功构建pET28a-PDIA3重组质粒,经IPTG诱导表达,超声破碎后通过镍柱亲和层析分离纯化,得到58 kDa的目的重组蛋白PDIA3,用牛胰岛素浑浊法测定其活性,以PDIA3为靶点,从商业化的化合物库中虚拟筛选小分子抑制剂,胰岛素比浊法进行抑酶活性测定。结果表明,纯化后的蛋白质二硫键异构酶a3(PDIA3)具有催化二硫键还原成巯基的能力,PDIA3的比活力为4.17U/mg,筛选到PDIA3小分子抑制剂化合物去甲氧基姜黄素和 41974954,且抑制效果与自身浓度成正比。
猜你喜欢
——一道江苏高考题的奥秘解读和拓展
中学生物学(2022年7期)2022-09-07
成都医学院学报(2022年4期)2022-08-19
天津医科大学学报(2021年4期)2021-08-21
江西农业学报(2021年4期)2021-04-20
现代临床医学(2021年2期)2021-03-29
中国金属通报(2021年20期)2021-03-11
中国化妆品(2020年6期)2020-07-22
三农资讯半月报(2020年11期)2020-06-21
商品与质量(2019年31期)2019-11-28
分析科学学报(2016年2期)2016-10-15
