
胃食管反流病分子生物学的研究进展
2022-12-24 02:41:56董茗茗柴秀坤刘学臣何云李艾迪蒋树林
中国比较医学杂志 2022年10期
董茗茗柴秀坤刘学臣何 云李艾迪蒋树林
(河北医科大学第二医院消化内科,石家庄 050000)
胃食管反流病(gastroesophageal reflux disease,GERD)是胃内容物反流至食管、口腔等多器官导致一系列临床表现和并发症的疾病[1]。 依据胃镜表现,可将GERD 分为非糜烂性胃食管反流病(nonerosive gastroesophageal refux disease,NERD)、反流性食管炎(reflux esophagitis,RE) 和Barrett 食管(Barrett’s esophagus,BE)三种类型。 据流行病学统计显示,以至少每周1 次的反流和/或烧心为标准,GERD 在东亚人群患病率为2.5%~7.8%,虽然低于世界范围内患病率13.3%,但也有逐年上升的趋势[2-3]。 反复胃内容物反流引起食管粘膜的糜烂、溃疡,若损伤因素长期存在还可能发生化生[4-5]。在GERD 患者中大约有10%~15%的BE 患者,并以每年约0.12% ~ 0.6% 的速度进展到食管腺癌(esophageal adenocarcinoma,EAC)[6-8]。 EAC 的5年总生存率仅约为10%~15%,而在过去三十年里EAC 的发病率稳步上升,在一些国家已经超过鳞状细胞癌,成为食管恶性疾病中最常见的组织学类型[9-10]。 环境因素和导致基因组不稳定性的遗传倾向共同调节正常鳞状上皮-糜烂上皮-肠上皮化生-异型增生-腺癌的发生过程,既往国内外学者已经对其中的分子转化和进展机制进行了大量研究。本综述旨在描述GERD 发生及发展为EAC 过程中主要的分子生物学作用。
1 从鳞状上皮到肠上皮化生
1.1 遗传基因
COL3A1 基因位于染色体2q32.2,编码III 型胶原α1 链(COL3A1),胶原单体组成原纤维后可以聚集成纤维,为中空器官提供组织支撑结构。 在2009年,Asling 等[11]在GERD 显性遗传家族基因检测中发现COL3A1 与GERD 发生显著相关,且COL3A1在儿童和成人GERD 中均为疾病相关基因,男性患者食管活检组织中III 型胶原蛋白含量更高(P=0.03)。 此次研究为GERD 存在结缔组织薄弱成分这一概念提供分子支持,同时体现出性别在GERD的遗传风险中存在差异。 在后续研究中对36 个显示GERD 显性传播的家族进行全基因组基因分型和连锁分析,又确定了16 号染色体上的基因4-氨基丁酸氨基转移酶(aminobutyrate aminotransferase,ABAT)内含子中的一个单核苷酸在儿童中具有显著的遗传相关性,并且在动物实验中,通过选择性ABAT抑制剂减少了约57%暂时性食管下括约肌松弛(TLESRs),胃内容物反流也减少70%左右。Jirholt 等[12]进行的两项双胞胎研究表明,GERD 的遗传率约为30%。COL3A1 和ABAT基因的发现不仅对GERD 的发生机制做出了贡献,还为遗传性危险因素增添了新成员,但ABAT等位基因在瑞典成人病例对照队列中并未发现是GERD 遗传的危险因素,应该在不同人种显性遗传的家族中进行更多研究。
1.2 白细胞介素-1 基因簇与GERD
白细胞介素-1(interleukin-1,IL-1)基因簇位于2q12 染色体,由IL-1A、IL-1B和IL-1RN3 个相关基因组成,分别编码促炎细胞因子IL-1α 和IL-1β 及其内源性受体拮抗剂IL-1Ra[13]。 IL-1B 在启动子-511 和-31 区域有两个双等位基因多态性,分别代表C/T和T/C转换,研究表明,这两个区域的基因几乎处于完全连锁不平衡状态,即基因并非完全随机组成单体型,有些基因总是较多的在一起出现,从而使某些单体型在群体中出现频率较高[14]。 而在IL-1RN的内含子中含有可变数量的串联重复多态性(VNTR),串联重复指DNA 短序列在特定的染色体位点以头到尾的方式重复,每一个都是遗传等位基因,共包括5 种不同的等位基因,即IL-1RN*1(4 次重复)、IL-1RN*2(2 次重复)、IL-1RN*3(5次重复)、IL-1RN*4(3 次重复)和IL-1RN*5(6 次重复)。 有研究称IL-1B-511*T等位基因或IL-1B-31*C 等位基因的存在对GERD 的发展具有保护作用,不仅可能导致胃炎从而破坏壁细胞,还能诱导胃体萎缩减少胃泌素刺激,导致胃酸分泌减少从而降低GERD 的严重程度[15]。IL-1RN*2 的存在与高IL-1Ra 和低IL-1β 释放有关,IL-1β 和IL-1Ra 之间的平衡是局部组织炎症程度的决定因素,已经证明在胃炎、胃癌、肠化生等多个疾病中起重要作用。Akçil 等[16]对不同程度的食管炎患者进行基因分析,发现在幽门螺杆菌感染的情况下与对照组相比,RE 患者IL-1B-511*TT基因型频率明显低于NERD,但在IL-1B-31 和IL-1RN方面没有差异。 同样,Ghoshal 等[17]在一项对144 名GERD 患者和368名来自印度的健康对照的研究中也证明了IL-1B-511*T/IL-1RN*1 单倍型受试者患GERD 的风险降低。 但与前两项研究结果相反的是,台湾一项研究表明IL-1B-511*T/T(P=0.034)、IL-1B-31*C/C(P=0.031)基因型和IL-1B-511*T(P=0.044)和IL-1B-31*C(P=0.040)等位基因与食管炎复发风险增加相关。 为了明确IL-1 单核苷酸多态性(single nucleotide polymorphisms,SNPs)及基因的单倍型在GERD 发生发展中究竟是保护因素还是风险基因,一项对白种人进行的前瞻性研究结果显示单倍型T(IL-1A-889)C(IL-1B-511)C(IL-1B-3953)L(IL-1RN)、C(IL-1A-889)C(IL-1B-511)C(IL-1B-3953)L(IL-1RN)与GERD 和RE 的发生风险增加相关,而T(IL-1A-889)C(IL-1B-511)T(IL-1B-3953)L(IL-1RN)可减少粘膜化生发生率,降低BE 的发生风险[18],但在此研究中没有分析被研究人员幽门螺杆菌感染情况,不能说明有无感染之间各种基因的差异。 综上研究,在GERD 患者中不同的单倍型可能参与从GERD 到RE 和BE 的进展,评估IL-1B、IL-1RN 及单核苷酸多态性是有必要的,以此发现有风险发展BE 和EAC 的患者。 但幽门螺旋杆菌的感染、人种区别也可能会对最终结果产生影响,具体作用机制暂不清楚,因此感染、GERD 发生发展及基因型之间的关系还需在不同人种间做大样本的深入研究。
1.3 蛋白酶激活受体-2
蛋白酶激活受体-2(protease-activated Receptor-2,PAR2)是一种G-蛋白偶联受体,存在于多种细胞中,如免疫细胞MCs、上皮细胞和嗜酸性粒细胞,并与细胞趋化、炎症、血管扩张等相关[14]。 PAR2 在食管上皮中表达,很容易与胃食管反流物(胃酸、胰蛋白酶)相互作用,参与GERD 的粘膜免疫发病机制。 在动物模型中,上皮暴露于反流事件后PAR2表达上调,可能通过引起紧密连接蛋白的重新分布而增加食管上皮的通透性,同时激活上皮细胞大量分泌IL-8 并引发促炎性粘膜免疫反应,导致上皮细胞间空间增大和上皮屏障进一步破坏,引起GERD的发生、发展[19]。 研究者认为GERD 患者中上皮内存在T 细胞和MCs 浸润,MCs 参与应激诱导的食管粘膜功能障碍,而PAR2 是潜在的中间因子。Winkelsett 等[20]、Keita 等[21]鉴定了人类食管上皮中PAR2 的表达,并证明了PAR2 介导的途径在GERD相关粘膜改变的发病机制中的功能重要性。 以上研究证明了GERD 发病机制中免疫介导粘膜炎症的假说,也说明需要更多的研究来充分了解基因、受体、传导通路的作用和具体的发病机制,帮助我们识别高危患者、确定治疗目标以帮助更多患者。
1.4 谷胱甘肽-S-转移酶与GERD
谷胱甘肽-S-转移酶(glutathione s-transferase,GST)是II 期解毒酶,存在于许多物种和组织中,包括人类胃肠道的上皮组织中,可以催化谷胱甘肽对多种疏水亲电试剂的亲核攻击,产生毒性更小、水溶性更强的化合物,在保护细胞免受氧化应激产物的伤害方面及防止肿瘤发展中具有重要作用。 在正常食管上皮组织中,GSTP1 是GST的主要亚型,GSTP1 编码区内核苷酸+313 处的A转换为G,104密码子将从ATC变为GTC,这将导致GST 活性降低[17]。GSTP1*B的等位基因频率在正常东方人群中为23%,在白种人中为30%[22]。 有研究发现,GSTP1*B等位基因与GERD 易感性及BE 发生相关,BE 患者中GSTP1 *B等位基因携带者相比GERD 患者(OR 2.10,95% CI:0.99~4.44)和健康对照组(OR 2.56,95%CI:1.30~5.05)发现频率显著增加。 国内一项研究发现变异GSTP1 基因型A/B在RE 患者、NERD、健康对照组中出现频率分别为40%、25%和22%,具有显著差异(P<0.05);且在GSTP1 基因型变异的受试者中,RE 发生风险增加2.42 倍(OR=2.42;95%CI:1.22~4.80)[23]。 以上研究结果提示我们GSTP1 基因多态性可能是RE 发病机制的高易感性因素之一。
1.5 G 蛋白β 亚单位基因与GERD
G 蛋 白 β 亚 单 位 基 因(guanine nucleotide binding protein(Gprotein),beta polypeptide 3,GNB3)位于染色体12p13。 G 蛋白在信号传导过程中起“分了开关”作用,介导对酸、神经递质和调节食管感觉功能的某些体液因子的反应,而在G 蛋白亚单位中,β 亚单位是α 亚单位以及某些信号转导受体和效应器的重要调节因子。 有研究表明食管症状严重程度与反流负荷之间并无直接相关性,部分原因可能是GERD 患者对食管症状的感知存在差异,而这一过程与基因决定感知信号转导相关。 2018年的一项研究表明,在接受动态反流试验的患者和无食管症状的对照组之间,GNB3 内3 个单核苷酸多态性的等位基因Rs2301339(P=0.040)、Rs5443(P=0.011)和Rs5446(P=0.016)之间存在显著差异,而且与显性纯合子相比,尽管食管酸暴露水平和症状反流相关程度相似,但是隐性等位基因携带者Rs2301339*A、Rs5443*T、Rs5446*T的反流症状更重、心理健康相关生活质量和贝克抑郁量表得分更差[24]。 在另一项病例对照研究(363 名白人GERD 患者和373 名健康对照)结果中,GNB3825*TT可预测G 蛋白活化增强,从而增加细胞或生理反应;GNB3825*CC使信号转导反应减弱。 最近有研究发现希腊、日本人群中的炎症基因多态性GNB3与功能性消化不良中的上腹部疼痛综合征患者相关。 因此表明GNB3 基因的多态性可能介导食管粘膜对酸性递质的反应,导致患者出现反酸、烧心、疼痛等多样的临床表现,也为GERD 患者对治疗的不同敏感性提供理论依据。
2 从肠上皮化生到腺癌
2.1 环氧合酶-2
环氧合酶-2(cyclooxygenase-2,COX-2)基因位于1q25.2-q25.3,编码COX-2 蛋白,在细胞因子、炎症介质、内毒素等诱导刺激下催化花生四烯酸转化为前列腺素。 COX-2 可以促进细胞增殖、降低凋亡率,还能刺激血管生成,因此在炎症和肿瘤发生中发挥着重要作用[25-26]。 在体外实验中,反流物可刺激诱导食管上皮细胞过表达COX2,COX2 的表达与胃肠粘膜的化生、癌变相关,而应用COX2 抑制剂后降低了大鼠模型中EAC 的发生风险。 基于体外实验的结果,研究人员开展了一项病例对照研究,结果发现COX-2-8473*C等位基因的存在可能使个体易患EAC,但与BE 或GERD 的发生无关。 也有研究表示COX-2 启动子区域的两种多态性-765C/G、-1195A/G与EAC 发生相关,亚洲人群中-765*C等位基因可能是EAC 发生的危险因素。 COX-2作为预测肿瘤进展的可能生物标志物及选择性抑制COX-2 作为BE 的防治方法受到了广泛关注。 在最近的随机多中心研究中,研究者发现应用大剂量的埃索美拉唑和阿司匹林可以改善BE 患者的预后[27]。
2.2 表皮生长因子
表皮生长因子(epidermal growth factor,EGF)基因位于4q25,表皮生长因子(EGF) 及其受体(epidermal growth factor receptor,EGFR)属于酪氨酸激酶受体家族,结合后会发挥促进有丝分裂的作用,在粘膜损伤的保护和修复中具有重要作用,基因突变时会表达异常破坏上皮屏障的完整性,病理水平与胃肠道肿瘤发生相关。 Cheung 等[28]对309例EAC 患者和275 例健康对照者的DNA 样本进行EGF基因分型,发现病例组中EGF变异(A/G或G/G)较对照组有显著差异(P=0.02),且与A/A基因型相比,G/G突变与GERD 患者发生EAC 风险显著增加相关(OR=9.7;95%CI:3.8~25.0;P<0.001),同时G/G基因型与GERD 存在高度显著的交互作用(P<0.001)。 Lanuti 等[29]也提出与对照组相比,EGF-A61*G/G基因型会导致EAC 发生风险增加约2 倍(OR=1.81;95%CI:1.2~2.7),且在伴有BE的EAC 患者亚组中更高(OR=2.18;95%CI,1.3~3.7)。 2012 年,Menke 等[30]为确定EGF基因多态性与RE、BE、EAC 之间的关系,对荷兰白种人进行了队列研究,结果表示与A/G(P=0.008)和A/A(P=0.002)组相比,G/G基因型的EGF表达显著降低。EGF表达降低与RE(OR=2.6;95%CI:1.3~5.2)、BE(OR=3.0;95%CI:1.5~6.2)和EAC(OR=4.1;95%CI:1.8~9.7)发生风险增加相关。 体外研究发现,用酸性胆汁盐处理食管细胞可激活EGFR信号,而且在22.2%~35%的BE 和46.5%~80%的EAC 患者中发现EGFR 蛋白表达增加[31-32]。 因此研究者认为EGFR 信号的异常激活是由EGFR 蛋白及其配体TGF-α 和EGF 的过度产生引起,在食管炎患者中EGF 与PG/COX-2 和PPARγ 系统相互作用,提出应用EGFR 抑制剂和PPARγ 激动剂可以用来治疗进展为BE 的慢性食管炎。
上述研究发现EGF的单核苷酸多态性也在食管粘膜上皮化生到异型增生以及随后转化为腺癌的进展中有一定作用,其中最主要的基因G/G突变,基因突变后遗传性EGF 表达降低导致的粘膜保护减少可促进食管肿瘤的发展。 因此基因分型可以用来识别GERD 和BE 中的高危患者,且EGFR的激活也与BE 发生相关,阻断此信号传导途径可以用于治疗BE。
2.3 TP53 基因
TP53 是位于染色体17p13 的抑癌基因,又称原癌基因,TP53 基因编码的p53 蛋白在调控细胞分裂、增殖、凋亡及DNA 修复中有非常重要的作用。许多研究报告了TP53 基因突变是迄今为止与癌症亚型(尤其实体瘤)关系最密切的基因[33],该突变已被证明在绝大多数食管腺癌中发生,并且是Barrett 食管向腺癌进展的潜在生物标志物[34]。 目前研究认为TP53 在体内存在两条途径,一种是早期突变导致TP53 失活,引起p16 失活,最后激活ERB-B2 原癌基因;另一种途径是TP53 突变,随后全基因组加倍,最终基因组不稳定和原癌基因被激活[35]。TP53 基因突变在进展为高度异型增生及EAC 的BE 患者中数量显著增加,使BE 高度发育异常进展风险增加13.8 倍(95%CI,3.2~61.0;P<0.001)[36-37]。 研究表明,在酸性反流物刺激下,胃食管反流病患者中由自由基诱导的脂质过氧化和COX2 酶产生的反应性异葡萄糖苷(reactive isolevuglandins,isoLG)会与p53 结合导致p53 活性被抑制[27]。
2.4 基质金属蛋白酶
基质金属蛋白酶(matrix metalloproteinases,MMPs)属于锌依赖性内肽酶,在1962 年首次确定为胶原蛋白水解酶之一, 参与细胞外基质(extracellular matrix,ECM)的重塑 。 据报道基质金属蛋白酶参与调节肿瘤进展过程中的关键事件,如细胞存活和侵袭、转移发展和血管生成以及恶性转化等[38]。 在大约MMP的30 种亚型中,小鼠GERD模型中MMP3 和MMP9 的水平上调,MMP1、MMP2、MMP7 和MMP9 的表达与BE 和EAC 相关。 由于发现MMP基因多态性与EAC 风险增加相关,Cheung等[39]对309 名EAC 患者和279 名健康患者进行基因分型检测,确定MMP-1*1G/2G(OR=3.2,95%CI;2.0~5.1,P<0.001)和、MMP-3 *6A/5A(OR 1.8,95%CI:1.1~2.7,P= 0.01)在GERD 患者中与EAC 风险增加独立相关,且与不同严重程度GERD 患者风险不同,因此提出MMP-1*1G/2G、MMP-3*6A/5A的基因多态性与GERD 病史相关,并且协同增加了EAC 的发生。 另一项横断面研究也提出,尽管BE 患者组中没有上皮恶变,但与对照组相比BE 患者MMP-9 和MMP-3 表达增加。 研究者通过动物实验提出通过抑制TLR4/NF-κB 信号通路,食管粘膜组织中的炎症因子(MMP3、MMP9)水平下调,食管粘膜组织损伤减轻。 无论是动物实验还是病例对照研究,都证明了MMP基因多态性与GERD、BE、EAC 相关,不同的研究对表达调控的机制有一定帮助。
2.5 CCND1 基因与细胞周期蛋白D1
细胞周期蛋白D1(cyclin D1)由位于人类11q13 染色体的CCND1 基因编码,与细胞周期蛋白依赖性激酶(cyclin-dependent kinase,CDKs)结合形成复合物后驱动细胞周期从G1 期进展到S 期增殖。CCND1 基因在外显子4(G870A)中表现出单核苷酸多态性,CCND1*A/A 基因型的存在与GERD、BE 和EAC 的风险增加相关。 研究人员发现与无症状患者相比,A/A基因型与GERD、BE 和EAC 的发生风险增加3~6 倍,而且15 名(20%)GERD 患者、14 名(11%)BE 患者和14 名(25%)EAC 患者中细胞周期蛋白D1 的过度表达也存在显著差异(P=0.001)。 在另一项小型病例对照研究中,进展为食管腺癌的BE 患者中67%的食管活检组织染色中发现了细胞周期蛋白D1,而在未进展的BE 患者中比例只有29%。 因此具有CCND1*A/A基因型的个体对GERD、相关疾病有易感风险,同时细胞周期蛋白D1 的过度表达可以用于鉴别有恶性肿瘤发生风险的BE 患者。 但在有关研究中并未发现基因型和细胞周期蛋白D1 过度表达之间存在相关性[40],也有研究者提出细胞周期蛋白D1 的表达似乎不能预测BE 患者的肿瘤进展,CCND1 基因型与细胞周期蛋白D1 的表达在GERD 发生发展的作用仍然存在争议。
2.6 尾侧相关同源盒转录因子2
尾侧相关同源盒转录因子2(caudal-related homeobox transcription factor 2 ,CDX2)是一种特异性转录因子,正常情况下仅在肠道表达,指导和维持肠细胞的分化、促进肠道发育[41]。 既往研究显示胆汁酸处理后正常胃黏膜上皮细胞化生,CDX2、粘蛋白2(mucin 2,MUC2)表达增加[42]。 在最近的一项研究中,在用胆汁反流建立的大鼠模型和酸性培养基中的人食管鳞状上皮细胞中,检测发现Krüppel-like factor 5(KLF5)、CDX2 及肠道标志物表达均增加。 同时将KLF5 表达载体转染食管上皮细胞可促进其向柱状细胞的分化同时CDX2 表达增加,基因敲除KLF5 后可阻断CDX2 等相关蛋白的表达[43]。 在另一项研究中,小鼠模型中的食管鳞状上皮暴露于酸和胆汁酸时,炎症和组织损伤激活信号通路,如sonic hedgehog、bmp4 和核因子κB 表达,并下调Notch 信号,最终导致Sox9(诱导柱状分化)和Foxa2、Cdx1 和Cdx2(诱导肠道分化)的表达增加[44]。 因此CDX2 目前被认为是层状鳞状上皮化生为柱状上皮的转录调节因子之一,但具体通路的调节与多因子相关,且目前研究多为动物实验,尽管这些结果提示我们新的生物标记物,但在临床广泛推荐前仍需进行大规模前瞻性临床试验。
3 其他与GERD 发生、发展相关研究
2020 年Westra 等[45]的研究结果发现,F 单倍体在EAC、BE 和GERD 患者中的表达比例分别为34%、27%、23%,提出F 单倍体组是EAC 的危险因素(OR1.5;95%CI:1.03 ~ 2.19,P= 0.03)。 Lam等[46]也证明FOXF1 rs9936833*C、MHCrs9257809* A、IGF1rs6214、GHrs6898743、BARX1 及ADAMTS17 有可能和GERD 发生有关。 同时基于对两个多代BE 和/或EAC 家族的测序,Verbeek 等[47]也已经确定了两个可能致病的基因变体VSIG10L和MSX1。 然而,以上研究中发现的相关基因只是初步的观察结果,还需进一步在人群中进行大量研究加以验证。
4 小结与展望
GERD 是一种与多个炎症和代谢生物标志物循环水平相关的疾病,症状反复发生降低了患者的生活质量,随着基因组学的快速发展,目前已经发现COL3A1、ABAT、IL-1 基因簇、PAR2、COX-2、GNB3 等基因与GERD 发生风险相关,同时TP53基因突变及EGF、MMP、CCND1、cyclin D1、CDX2、VEGF 的单核苷酸多态性促使部分个体向BE 及EAC 进展(如图1)。 这些研究进展丰富了疾病在生物学方面的理解,为疾病的预防及诊疗提供了新思路。 但是部分信号通路主要是在体外实验中研究,未来的研究应该从健康患者向疾病状态的转化去深入了解,同时在不同人群中重复,希望建立生物标记物为早期发现、临床干预及靶向治疗提供可靠依据。
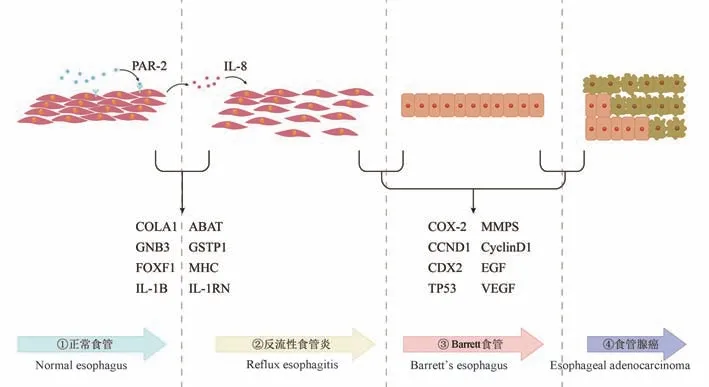
图1 GERD 发生发展中主要基因改变图示Note. COL3A1, ABAT, IL-1 gene cluster, COX-2, GNB3 and other genes are related to the occurrence of reflux esophagitis, while TP53 gene mutation and EGF, MMP, CCND1, CDX2, VEGF are involved in the progression of Barrett’s esophagus to esophageal adenocarcinoma.Figure 1 Illustration of major genetic alterations in the development of GERD
猜你喜欢
智慧健康(2021年17期)2021-07-30 14:38:32
妈妈宝宝(2019年10期)2019-10-26 02:45:28
现代检验医学杂志(2016年5期)2016-08-20 03:16:54
法医学杂志(2015年4期)2016-01-06 12:36:40
中国癌症杂志(2015年4期)2015-12-09 03:15:52
健康管理(2015年3期)2015-11-20 18:22:36
实用手外科杂志(2015年3期)2015-08-27 01:53:24
中国中医药现代远程教育(2014年23期)2014-03-01 04:33:34
中国中医药现代远程教育(2014年17期)2014-03-01 04:29:22
分子诊断与治疗杂志(2013年3期)2013-07-08 02:17:11
