
胶原蛋白改性聚乳酸-羟基乙酸载药纳米纤维膜的制备及其性能
2022-12-23 07:09:24吴焕岭谢周良孙万超康正芳徐国华
纺织学报 2022年11期
吴焕岭, 谢周良, 汪 阳, 孙万超, 康正芳, 徐国华
(1.盐城工学院 纺织服装学院, 江苏 盐城 224051; 2.盐城市权航科技有限公司, 江苏 盐城 224056; 3.盐城创能新屏蔽材料有限公司, 江苏 盐城 224043)
聚乳酸-羟基乙酸共聚物(PLGA)具有优异的生物相容性和体内降解性,在体内可通过水解作用全部降解,且具有非常好的可塑成型性,能够用于药物载体材料、医用组织工程材料等领域。为了实现较高且稳定的力学性能,一般采用中、高分子质量PLGA材料制备组织工程支架材料。与此同时,这些支架材料的降解速率缓慢,但在体内降解周期较长,有些可长达6个月以上[1-2],在体内长期留存将产生潜在机体排异和组织炎症等副作用。另外,由于PLGA较强的疏水性,将其用于药物传递材料时常常会出现药物释放速率慢、药物累积释放量低等问题。
基于构效关系与表界面作用理论[3-5],通过复配调控方式将不同材料的分子链从分子水平进行重排,能够减弱高聚物大分子之间的作用力,改变材料的亲疏水性和降解性,也会对细胞在生物材料表面的黏附增殖性能产生较大影响。有研究表明,低分子质量与高分子质量的聚己内酯(PCL)通过共混静电纺丝的方式,能够有效改善高分子质量PCL纳米纤维膜的降解性[6]。也有研究显示:借助静电纺丝技术制备PLGA、胶原蛋白复合纤维膜,可用于修复硬脑膜支架材料[7]、骨材料[8]和缝线材料[9],能够显著提高PLGA材料的降解速率。原因在于胶原蛋白的引入能够提高PLGA的亲水性和生物相容性[10],易于水分子的渗透和降解反应的发生。
在药物载体应用方面,将药物载入特定的有机或无机材料中,再置于肿瘤部位,能够在肿瘤部位富集较高浓度的药物,实现药物的长效、精确输送,降低给药频率,且避免口服药的首关效应及全身性毒副作用[11-13]。近年来,静电纺丝技术飞速发展,由于该技术所制备的纳米纤维具有比表面积高、孔隙率高等优点,已经成为制备纳米纤维最简单高效的技术之一,并广泛用于药物缓释材料和组织工程支架材料等领域[14-15]。基于以上分析,为改善PLGA基药物载体的降解性能和释药性能,本文以PLGA为基材,以胶原蛋白(Col)为改性材料,以盐酸阿霉素(DOX·HCl,文中均简写为DOX)为药物模型,利用静电纺丝技术制备PLGA/Col/DOX复合载药纳米纤维膜,作为乳腺癌局部治疗药物缓释材料使用,并对其结构和性质进行表征与分析。
1 实验部分
1.1 实验材料
材料:聚乳酸-羟基乙酸(重均分子质量为120 000~160 000),济南岱罡生物工程有限公司;磷酸缓冲盐溶液(PBS,pH值为7.4),南京森贝伽生物科技有限公司;透析袋(截留相对分子质量为2.5×104),上海吉至生化科技有限公司;盐酸阿霉素(纯度为98%)、牛跟腱I型胶原蛋白、六氟异丙醇(HFIP,纯度为99.5%),上海麦克林生化科技有限公司;噻唑蓝(MTT)胰酶,西格玛奥德里奇(上海)贸易有限公司;二甲基亚砜,国药集团化学试剂有限公司;DMEM 高糖培养基、胎牛血清,Hyclone公司;小鼠成纤维细胞L929,中国科学院。
1.2 静电纺丝溶液的配制与纤维制备
1.2.1 DOX、PLGA和Col溶液的制备
以HFIP作为DOX、PLGA、Col的溶剂,分别配制质量浓度为0.1 g/mL PLGA溶液150 mL、0.01 g/mL DOX溶液10 mL和0.1 g/mL Col溶液20 mL。
1.2.2 纺丝液的复配
将不同溶液组分按照表1所示的体积比进行混合,并充分搅拌均匀,分别得到PLGA、PLGA/Col、PLGA/DOX、PLGA/Col/DOX 4种纺丝溶液[16]。

表1 纺丝液的复配组分Tab.1 Composition of spinning solution
1.2.3 静电纺纳米纤维膜的制备
将配制的纺丝液通过DP30-S型静电纺丝机进行纺丝。纺丝条件为:温度25 ℃,相对湿度45%,电压18 kV,接收板与喷丝头距离14~16 cm,溶液流速为1.2 mL/ h,接收基材为铝箔纸。每个样品纺制平行样3份,每份纺丝时长均为1 h[16]。
1.3 测试与表征
1.3.1 纤维形貌观察
利用Nova Nano-450型场发射扫描电子显微镜观察纳米纤维膜样品的外观形貌,测试前先将纳米纤维膜连同其所附着的锡箔纸一同裁剪成0.5 cm × 0.5 cm尺寸大小,用导电胶粘贴到专用载物台上,并进行喷金处理45 s。
1.3.2 力学性能测试
将待测纳米纤维膜从烘箱中取出,先裁成2 cm×7 cm规格,再揭除锡箔纸只留下纳米纤维膜,采用WDS-20型断裂强力测试仪测试试样的力学性能。测试环境为:温度20 ℃,相对湿度65%。样品测试参数为:夹持长度5 cm,拉伸速率25 cm/min。
1.3.3 接触角测试
将纳米纤维膜连同其所附着的锡箔纸一同裁剪成2 cm × 2 cm规格,平铺并固定在JCY-2型接触角测试仪工作台上,测试并读数。测试参数:液滴体积3 μL,采用去离子水。
1.3.4 化学结构表征
采用NEXUS-670型傅里叶变换红外光谱仪对纳米纤维膜进行化学结构测试,扫描范围为4 000~500 cm-1。
1.3.5 热分析
采用STA-449C型TG-DSC同步热分析仪分别对不同纳米纤维膜进行降解性能测试。测试环境为N2气氛,升温速率为10.0 ℃/min,温度范围为50~800 ℃。
1.3.6 体外降解实验及降解性能测试
利用质量损失法分别对胶原改性前后的纳米纤维膜进行降解性能测试。具体过程为:首先,精确称量纳米纤维膜25 mg并装入透析袋中封口,再置于50 mL离心管底部,随后向离心管注入25 mL PBS缓冲溶液,并设置3份平行样;然后,将试样置于恒温水浴振荡摇床中,在37 ℃、100 r/min的条件下分别恒温振荡降解0、3、7、15、22和30 d;最后,称量降解后的质量,通过下式计算质量损失率:
式中:m0为纳米纤维膜降解前的质量,mg;mt为纳米纤维膜降解后的质量,mg。
1.3.7 体外释药性测试
将恒温振荡摇床预先设置为37 ℃、100 r/min,备用。精确称量Col改性前后的纳米纤维膜25 mg并装入透析袋中封口,再置于50 mL离心管底部,随后向离心管注入25 mL PBS缓冲溶液,并设置3份平行样。用封口膜将离心管密封完好,之后迅速置于恒温振荡摇床中。在一定的时间间隔从样品离心管中吸取2 mL待测溶液,并及时补充2 mL新鲜PBS溶液。将取出溶液避光保存,并在2 h内使用UV1 810 S型紫外分光光度计将取出的样品溶液在480.0 nm(盐酸阿霉素的最大吸收波长)处测试其吸光度(A),并结合DOX的标准曲线计算释药浓度(C)和药物累计释放量。
1.3.8 细胞相容性测试
参照ISO 10993《生物相容性测试标准(生物学评价)》和GB/T 16886—2022《医疗器械生物学评价》进行测试。通过PT-3502型酶标仪测试570 nm(甲瓒的最大吸收波长)下吸光度值来反映细胞在不同膜材料上的黏附增殖能力。
2 结果与讨论
2.1 微观形貌分析
采用场发射扫描电镜观察所制备的不同纳米纤维膜的表面形貌,结果如图1所示。可见,纤维均呈现出清晰的网状交织结构。PLGA、PLGA/Col和PLGA/Col/DOX复合载药纳米纤维膜的单根纤维直径均在200~500 nm范围内,但呈现出逐渐变细的趋势,表明相同质量纳米纤维膜的比表面积在逐渐增大。对比来看,PLGA纳米纤维形貌最均匀,PLGA/Col/DOX复合载药纳米纤维最不规整,出现了少部分粘连现象。总体上,PLGA与胶原蛋白以体积比3:1进行复合后,仍可达到均匀纺丝的效果。

图1 不同纳米纤维膜的扫描电镜照片Fig.1 SEM images of different nanofibrous membranes
2.2 力学性能分析
表2示出改性前后纳米纤维膜的力学性能测试结果。可知,未改性未载药PLGA纳米纤维膜的拉伸断裂强度和弹性模量在3个样品中均最高,分别为5.22与180.3 MPa,说明其发生单位形变所需要的力最大。PLGA/Col复合纳米纤维膜具有最高的断裂伸长率,为63.6%,说明胶原的加入能够改善PLGA纤维的形变能力。但随着药物的载入,纳米纤维膜的拉伸断裂强度、断裂伸长率和弹性模量等均发生明显下降。分析原因可能是DOX药物的分子质量小,其在一定程度上破坏了PLGA大分子的有序排列,因此,在材料使用时应根据使用性能的需要匹配相应的复配比例,以获取适当的强力性能。

表2 不同纳米纤维膜的力学性能Tab.2 Mechanical properties of different nanofibrous membranes
2.3 亲疏水性分析
图2示出改性前后纳米纤维膜表面的水接触角。可知,PLGA纳米纤维膜表面在未改性未载药前的水接触角为93.5°,显示出PLGA材料的疏水性;PLGA/Col(51.5°)和PLGA/DOX(54°)复合纳米纤维膜表面的水接触角在改性和载药后均降至50°左右,充分证实胶原蛋白或DOX的载入能够使PLGA纳米纤维膜表面呈现出较大程度的亲水性。水接触角降低最为明显的是PLGA/Col/DOX复合载药纳米纤维膜,其表面水接触角降至0°。该结果进一步说明,胶原蛋白和DOX的协同作用能够极大地促进强疏水性PLGA材料表面亲水性的显著改善,实现了PLGA材料疏水性向亲水性的转变。究其原因,与改性材料胶原蛋白分子上的强极性基团有关,也与DOX·HCl的分子结构与性质有关。尽管经过酸化的阿霉素水溶性有了大幅提高,但其在水中的溶解度仍然较低。在喷丝过程中伴随着溶剂的不断挥发,盐酸阿霉素小分子物质会发生不同程度地聚集,并附着在纳米纤维表面上,能够大幅增加纳米纤维表面微结构的粗糙度,使材料表面的亲水性增强,因此,水接触角减小[17]。随着纳米纤维膜亲水性逐渐增大,水分子更易润湿聚合物并进入其内部,促进PLGA大分子发生水解反应,并有助于提高纳米纤维载药体系的药物溶出速率。

图2 不同纳米纤维膜的水接触角Fig.2 Water contact angle images of different nanofibrous membranes
2.4 化学结构分析

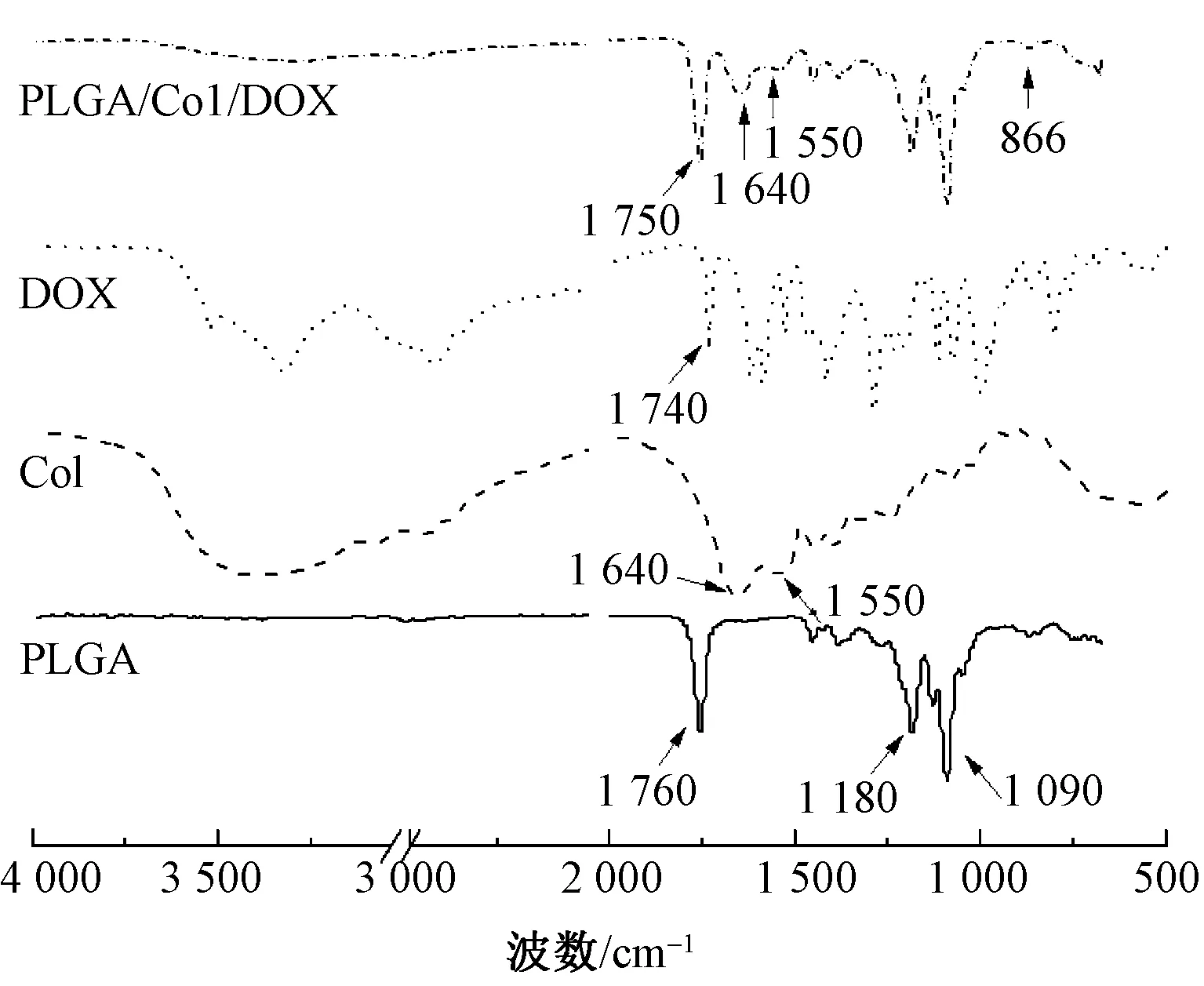
图3 不同材料的红外光谱图Fig.3 FT-IR spectra of different materials
2.5 纳米纤维膜的降解性能评价
2.5.1 热稳定性能分析
图4示出改性前后纳米纤维膜的热重曲线。可知,3种纳米纤维膜的热分解过程均始于250 ℃左右,并持续到400 ℃左右结束。虽然热降解起始温度非常接近,但后期的质量损失率、质量损失速率最大值对应的温度均有明显不同。PLGA纳米纤维膜的质量残留率仅约为1%,而PLGA/Col与PLGA/Col/DOX复合载药纳米纤维膜的质量残留率约为10%;PLGA纳米纤维膜的热质量损失速率最大值对应的温度约为375 ℃,而PLGA/Col与PLGA/Col/DOX复合载药纳米纤维膜均约为350 ℃,相差约为25 ℃。以上分析表明,经过复合改性后的纳米纤维膜更易发生热降解,说明胶原蛋白和药物小分子的加入能够加速PLGA的热降解历程[19]。
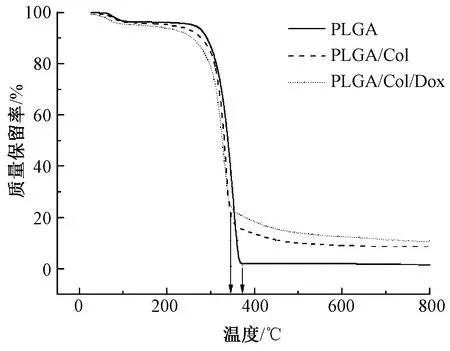
图4 不同纳米纤维膜的热重曲线Fig.4 Thermogravimetric curves of different nanofiber membranes
2.5.2 体外降解分析
图5示出胶原蛋白改性前后PLGA纳米纤维膜的降解情况。可知,胶原蛋白改性后的纳米纤维膜的质量损失率明显提高。在30 d的降解周期内,未改性PLGA纳米纤维膜的质量损失率仅为3.5%,而PLGA/Col复合纳米纤维膜的质量损失率增至19%。值得注意的是,2种纳米膜材料在降解前3 d均没有发生明显的质量损失。随着时间的延长,PLGA/Col复合纳米纤维膜的质量损失率快速增加,而未改性PLGA纳米纤维膜的质量损失率仍增加缓慢。该结果一方面与胶原蛋白复合改性使膜材料的亲水性提高有关,水分子更易进入纳米纤维内部,从而促进PLGA材料的降解。另一方面与PLGA本身的降解规律有关。现有研究表明PLGA材料的降解过程主要分为3个步骤:无规链段降解,此时分子质量下降较快,但无明显的质量损失;分子质量下降幅度减小,质量损失增加;形成大量溶于降解环境的低聚物并发生降解,质量损失增加明显[20-22]。这与本文实验结果一致,越到后期质量损失速率越快,且胶原的加入加速了降解历程。

图5 PLGA、PLGA/Col纳米纤维膜降解30 d后的质量损失率Fig.5 Weight loss rate of PLGA and PLGA/Col nanofiber membranes after 30 d of degradation
2.6 体外释药性能分析
图6示出DOX分别在PLGA纳米纤维膜和PLGA/Col复合纳米纤维膜中的药物释放情况。可见,PLGA纳米纤维膜和PLGA/Col复合纳米纤维膜的药物释放周期为120 h,二者的累积释药量分别为10与21.5 μg,说明经胶原蛋白复合改性后的纳米纤维膜的药物释放速率显著提升。PLGA纳米纤维膜在释放48 h时基本停止释放,究其原因PLGA是强疏水性材料,水分子较难穿越材料内部孔道进入到纤维内部,导致载药纤维内部的药物无法在水分子的作用下扩散到纤维外部,因此,所释放的药物主要来自于纤维表面附着的药物。而PLGA/Col/DOX复合载药纳米纤维膜在释药溶液中浸渍20 h后,释药速度明显加快,且48 h后仍在缓慢释药。主要原因在于胶原蛋白复合改性可使纤维的亲水性提高,水分子可穿越材料内部孔道进入到纤维内部,并将纤维内部的药物通过水分子的作用扩散到纤维外部。综上所述,经胶原蛋白复合改性后,PLGA由于亲水性的提高,纳米纤维载药体系的释药性得到改善,提高了药物持续释放作用和药物利用率。
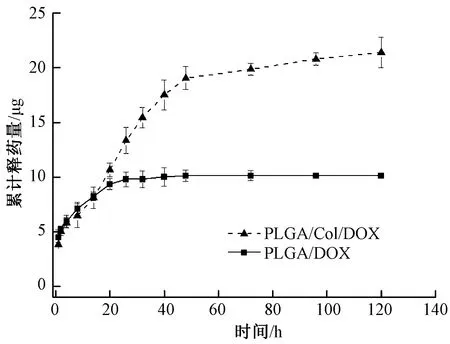
图6 PLGA/DOX、PLGA/Col/DOX载药纳米纤维膜的药物释放曲线Fig.6 Drug release curve of PLGA/DOX and PLGA/Col/DOX nanofiber membranes
2.7 细胞相容性评价
利用MTT法检测活细胞线粒体中琥珀酸脱氢酶的活力,可间接反映细胞在待测材料上的黏附增殖能力及共培养材料的细胞毒性。图7示出小鼠成纤维细胞L929分别与PLGA、PLGA/Col、PLGA/Col/DOX复合载药纳米纤维膜共培养1、3、5 d后的细胞增殖情况。可知,随着培养时间的延长,与3种纳米纤维膜共培养的细胞均显示出增殖趋势。其中,PLGA膜材料的细胞黏附增殖情况最差;胶原蛋白改性的PLGA/Col复合膜材料显示出最优的细胞黏附增殖性能,也说明材料对细胞毒性影响最低,细胞的活性最高;PLGA/Col/DOX复合载药纳米纤维膜材料显示出来的黏附增殖性能略低于PLGA/Col,与阿霉素抗肿瘤药物会对正常细胞产生一定毒性有关。该结果表明胶原蛋白复合改性通过改善PLGA的亲水性、电荷性等表界面性质,能够大幅提升PLGA材料的生物相容性,有利于细胞黏附增殖[3-4]。
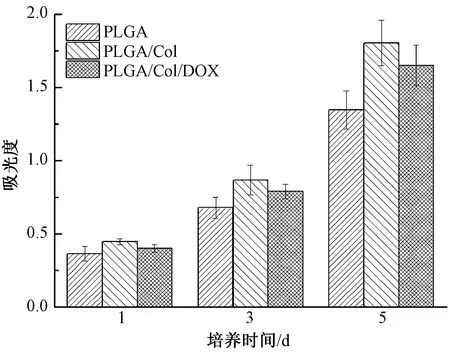
图7 L929细胞在不同纳米纤维膜上的黏附增殖能力Fig.7 Adhesion and proliferation of L929 cells on different nanofiber membranes
3 结 论
基于生物医用材料应具备一定的亲疏水性、可控降解性及细胞相容性等性质,本文采用胶原蛋白对聚乳酸-羟基乙酸共聚物(PLGA)进行复合改性以实现材料结构的重组和性能的改善,得到以下主要结论。
1)采用胶原蛋白对PLGA材料进行复合改性能够显著提高PLGA纳米膜表面的亲水性。PLGA与胶原蛋白(Col)以3:1的质量比进行复合后,制备得到复合纳米纤维膜的水接触角由93.5°降至51.5°。
2)采用胶原蛋白对PLGA材料进行复合改性能够改善PLGA降解速率过慢的缺陷,胶原蛋白改性前后纳米纤维膜30 d的质量损失速率分别为3.5%和19%,且改性后的载药体系的释药速率大幅提高。
3)采用胶原蛋白对PLGA材料进行复合改性能够提高PLGA材料的细胞相容性,有利于细胞黏附增殖。PLGA/Col/盐酸阿霉素(DOX)复合载药纳米纤维膜具有作为肿瘤局部治疗药物传递体系的应用潜力。研究策略可为生物材料及其表界面构建方法提供新的思路。
猜你喜欢
九江学院学报(自然科学版)(2022年2期)2022-07-02 02:33:28
云南化工(2021年7期)2021-12-21 07:27:36
Coco薇(2017年12期)2018-01-03 21:27:09
天然产物研究与开发(2016年6期)2016-06-05 10:29:30
天然产物研究与开发(2016年6期)2016-06-05 10:29:27
现代食品(2016年14期)2016-04-28 08:10:07
广东海洋大学学报(2015年4期)2016-01-13 08:39:40
燕山大学学报(2015年4期)2015-12-25 02:19:46
合成技术及应用(2015年3期)2015-12-11 08:36:27
化工进展(2015年3期)2015-11-11 09:07:30
