
Clonal Relationships among Enterococcus faecalis from Humans and Animal-Origin Foods in Xinjiang Characterized by Multilocus Sequence Typing
2022-12-22 09:08ZHANGXuelingYUANLixiaZHANGHuiminTIANFengweiNIYongqing
食品科学 2022年22期
ZHANGXueling,YUANLixia,ZHANGHuimin,TIANFengwei,NIYongqing,*
(1. School of Food Science and Technology, Shihezi University, Shihezi 832000, China;2. School of Food Science and Technology, Jiangnan University, Wuxi 214122, China)
Abstract: In this study, the population structure and evolutionary relationships of 59 Enterococcus faecalis strains isolated from breast milk, cheese, camel milk, mare milk and cold water fish from Xinjiang were analyzed by multilocus sequence typing (MLST). All the isolates were allocated into 12 sequence types, including three clonal complexes and three singletons. None of the sequence types were found to belong to high risk clonal complexes. Housekeeping gene split decomposition analysis indicated that gene recombination could be a major driving force in the evolution of E. faecalis.The results of minimum spanning tree indicated these E. faecalis strains were relatively weakly related to their geographical origin. Although eBURST and phylogenetic analysis demonstrated that the isolates from breast milk and cold water fish exhibited host specificity, E. faecalis clones closely related to them were still detected in breast milk, cheese, mare milk and camel milk. Taken together, E. faecalis from different hosts can adapt to new niches and spread via production practice and the food chain. Continuous monitoring is necessary to reduce the potential risk of zoonotic diseases
Keywords: Enterococcus faecalis; multilocus sequence typing; genetic diversity; clonal complexes; population structure
Enterococci are the generalist microbes highly prevalent inthegastrointestinaltractofhumansandanimals[1-2].These microorganismsalsoscatteredinsoil,water,plants,foodsand hospitalenvironments[3].Among enterococci,Enterococcus faecalisandE. faeciumarethemostprevalententerococcal speciesinvarioustypesofmeatproducts,cheeseandraw milkduetotheirresistanceagainstadverseenvironment[2].TheinterestinE. faecalishasgrownsignificantlymainly becauseit’scontributiontothesensorialproperties developmentandmicrobiotamodulationoffermented productsaswellasaccumulationofbioactivecompounds withpotentialbenefitsfortheconsumer’shealth[4-6].
However,asa“genetraffickers”,theuseofE. faecalisinthefoodindustryishighlycontroversialdue toitpropensitytoacquirevirulenceandantibioticresistance byhorizontalgenetransferbothwithintheirgenusandwith othermicroorganism[5,7-8].TheabilityofE. faecalistospread intodifferenthosttypesmightconstituteahumanhazard[9-10].Thedomesticationofanimalsandintensivelivestockfarming providedmoreopportunitiesforthespreadofpathogens betweenhumansandanimals[11].E. faecaliscancauseserious infectioninlivestockwhichwouldleadtolargeeconomic loss.Overuseandmisuseofantibioticsinfoodanimals hadledtotheemergenceofmultidrugresistantenterococci(vancomycin,gentamicin,quinupristinanddalfopristin)in animalsandmeat[12-13].And,theresistancegenesorresistant bacteriacouldbetransferredfromfoodanimalstohumans throughfoodchainsastheyaretightinterconnected[14-15].Atthesametime,thevirulenceandantibioticgeneof hospitalizedpatientscouldalsobedetectedinanimalsand foodviaplasmidsandtransposonstransmission[16-18].Over thepastthreedecades,overlappinginspecificsequence types(STs)andhigh-riskclonalcomplexes(CCs)between livestock,animalfoods,healthyhumansandnosocomial patientssuggestedthatinter-host-speciestransmission mayplayaroleinthespreadofmultidrugresistantE. faecalis[12,19-20].ItisdifferenttoquantifytheriskforE. faecalisofdifferenthosttypesinrelationtohumanhealth.So,elaboratingtherelationshipamongtheE. faecalisisolates fromdifferenthostsisbecomingincreasinglyimportant.
Nowadays,multi-locussequencetyping(MLST)is employedtocompareenterococcifromdifferenthosts[21-25].However,suchstudiesweretypicallylimitedtoclinical outbreaksandenvironmentsettingsand/orstudieslack strainsfromotherhosts,suchaslivestock,animalfoodand healthyindividuals.Xinjiangisthemainpastoralareaof China,wheredairyandmeatconsumptionplayanimportant roleinpeople’slives.Antibioticsareusedinlivestockto preventdiseasesandimproveproduction.Thus,foodanimals mightbepotentialreservoirsofresistancedeterminantsand virulencetraitstransferabletohuman-adaptedclustersby hostjumps[10,26].Themainpurposeofthisworkistospeculate therelationshipsamongclonalclustersofvariousorigins.To achievethisgoal,weappliedaMLSTmethodtoexplorethe populationstructureandtherelationshipamongtheisolates fromdifferenthosts.Thus,tofurtherelucidatewhether theanimal-adaptedandfoodoriginclusterswereobserved tospreadtohumansviadirectorindirecthuman-animal interconnectionorviatheconsumptionoforcontactwith animalproducts.
1 Materials and Methods
1.1 Materials and reagents
M17agar;TEbuffer(10mmol/LTris(pH8),10mmol/LEDTA);1 × TAEbuffer(40mmol/LTris,20mmol/Laceticacid,1mmol/LEDTA);the16SrDNA universalprimersofpolymerasechainreaction(PCR)was27F(AGAGTTTGATCCTGGCTCAG)and1492R(GGTTACCTTGTTACGACTT).
1.2 Instruments and equipments
SensoquestLabCyclerPCRsystemwaspurchased fromThermoFisherScientific;electrophoresisapparatus wasobtainedfromBio-Rad;BioNumericssoftwareV8.0 waspurchasedfromApplied-Maths,SintMaartens-Latem,Belgium;pyrosequencingwasperformedfromPersonal BiotechnologyCompanyGENEWIZ(Jiangsu,China).
1.3 Methods
1.3.1 Studysiteandsamplecollection
Thecollectionofsampleswereperformedin7regions ofXinjiangUygurAutonomousRegion(Tacheng,Bole,Yili,Altay,Kashgar)andGansuprovince(Zhangye,Gannan)in westernChina.Studysamplescomprisedofmaremilk,camel milk,breastmilk,coldwaterfishandtraditionalhandmade cheesemadefromcowmilk.Thetraditionalhandmade cheesewasmadeinanold-fashionedwaybylocalherdsmen,rarelyaddingadditionalstarter.Thecoldwaterfishlivedin EerqisiRiverinAltayregionofnorthernpartofXinjiang.AquaticenvironmentofEerqisiRiverhasnotbeenaffected bydifferentanthropicactivities,andmorethan10indigenous wildcoldwaterfisheswereinhabitedinthisriver.Breast milksamplesfromGansuprovincewerealsocollectedasthe controlgroup.
1.3.2 Strainsisolationandspeciesidentification
Serialdilutionsofthesupernatantsderivedfrom samplescollectedwereinoculatedontoM17agarwithtwo independentbiologicalreplicatesat37 ℃ for24hours.FivecoloniesperplatewithmorphologysuggestiveofEnterococcuswerestreakedforpurityandculturedinliquid media.Microscopewereemployedtoverifypurityand purecultureswerestoredat-80℃in25%(V/V)glycerol.1mLovernightcultureswereusedtoDNAextractusing themethodsdescribedbySingh et al.[27].Thefinalspecies identificationwasconductedbasedon16SrRNAanalysis with16SrDNAuniversalprimers27Fand1492R.ThePCR ampliconsweresequencedbytheGENEWIZBiotechCompany(Jiangsu,China)andthensubjectedtoBLASTanalysis.
1.3.3 MLSTscheme
SevenhousekeepinggeneswereselectedforMLST analysis[28],including:aroE(shikimate5-dehydrogenase),gdh(glucose-6-phosphatedehydrogenase),gyd(glyceraldehyde-3-phosphatedehydrogenase),gki(putativeglucokinase),pstS(phosphateATPbindingcassettetransporter),xpt(shikimate5-dehydrogenase)andyqiL(acetyl-coenzyme Aacetyltransferase)(Table 1).Allgeneshavelowratios ofnonsynonymoustosynonymousmutationsandahigh Simpson’sindexofdiversity.Afinalconcentrationof 0.2μmol/Lwasusedforeachprimerinthereactionmixture.ThePCRreactionmixtureconsisted12.5µLof2×PCR MasterMix(Vazyme,Beijing,China),8.5µLofnucleasefreewater,0.5µLofeachcoupleofprimersand3 µL oftemplateDNA(30ng/µL).EachPCRcyclingprofile consistedofacycleof94℃for10 min(pre-denaturation),followedby35cyclesof94℃for1min(denaturation),and 72℃for2min(extension),andfinally1cycle72℃for 4min(extension).Theannealingtemperatureofgkiwas 53℃,andtheannealingtemperatureofgdhandaroEwas 57℃.Therestswere52℃.ThePCRproductwasverified in1.2%agarose,andthenitwassequencedinGENEWIZ BiologicalScience&Technologycompany(Jiangsu,China).

Table 1 Primer sequences used for the amplification of housekeeping genes
1.3.4Computationalanalysis
Foreachgene,adistinctallelenumberwasassigned toeverydifferentsequence,inlinewiththeE. faecalisMLSTdatabase(http://efaecalis.mlst.net/),andeachunique combinationofsevenallelenumberswasallottedaST.New STsidentifiedinthisstudyweredepositedintheMLST database.Thephylogeneticanalysiswasperformedusing BioNumericssoftwareV8.0(Applied-Maths,SintMaartens-Latem,Belgium)bytheunweightedpair-groupmethod witharithmeticaverages(UPGMA)andthecategorical coefficientofsimilarity.Theminimum-spanningtree analysiswasconstructedwithPrims’salgorithmembedin theBioNumericssoftwareaccordingtoisolationsources andregions.Thenumberofpolymorphicsites,nucleotide diversity(π),G+Ccontent,Tajima’sDvalue,andthe nonsynonymoustosynonymousnucleotidesubstitutions ratio(dN/dS)werecalculatedwiththesoftwareDnaSpv5.0.Thegroupsofstrainsthatshareaminimumoffiveoutofthe sevenalleleswereplacedinaCC.TheeBURSTanalysiswas achievedtodetermineCCsandtofurtherinfertheevolutionary relationshipbetweenstrains.Split-tree v4.0softwarewasusedto evaluatetheimpactofrecombinationeventsonphylogeny,and splitdecompositionanalysiswasconstructed.APhi-testattached tothissoftwarewasusedtodetectwhethertherewassignificant recombinationinalleles.Thelinkageequilibriumbetween alleleswascalculatedwithLIAN3.0andthestandardizedindex ofassociationwasdetected.
2 Results and Analysis
2.1 Recovery of Enterococcus isolates
Totally,59representativeE. faecalisisolateswere retrievedfromthesamplesinvolvedinthisstudy.These includedtwentyisolatesfromhuman-origin(breast milk=20),thirty-nineisolatesfromanimalfood-origin(camelmilk=2,maremilk=4,coldwaterfish=15and cheese=18).Thedistributionoftheisolationsourcesand regionsisshowninFig.1.
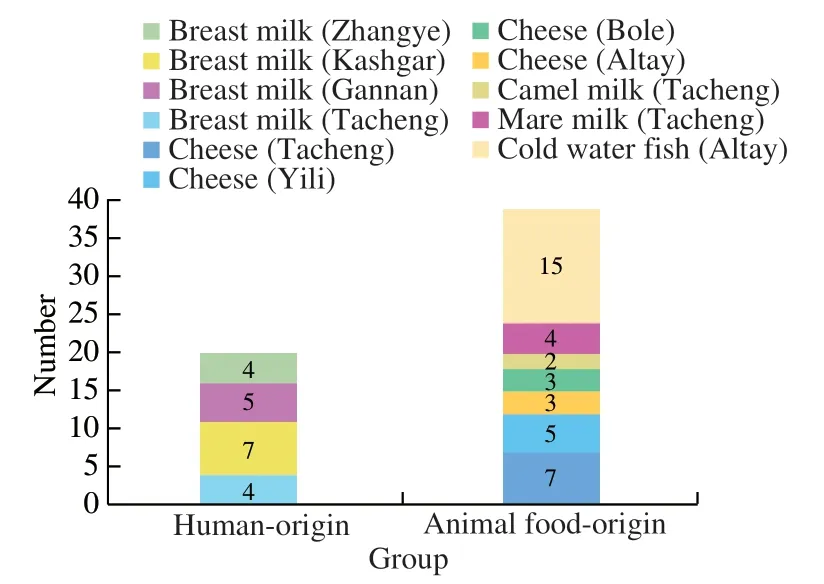
Fig. 1 Food sources and geographical origins of the 59 E. faecalis strains
2.2 MLST sequence analysis
Allsevenhousekeepinggenesweresuccessfully amplifiedfor59E. faecalisisolatesandallelicvariation weredescribedinTable2.Thenumberofallelesforeach locirangedfrom4(gyd)to7(pstS)andthenumberof polymorphicsitesrangedfrom4(xpt,psts)to10(aroE).Theaverageguaninecytosinecontentofthe7genesvaried between38.5%(gdh)and49.4%(gki),whichwassimilar withthe37.65%observedinE. faecalisATCC19433 genome.Theaveragepairwisenucleotidediversity(π)was rangingfrom0.00214(xpt)to0.00716(gki)pergene.The dN/dSvalueforthe7genesvariedbetween0.00391(gdh)and0.81104(yqiL)werealllessthan1.Moreover,Tajima’sDvaluewascalculatedforeachgeneandrangedfrom-0.64324(gyd)to1.83869(gki).
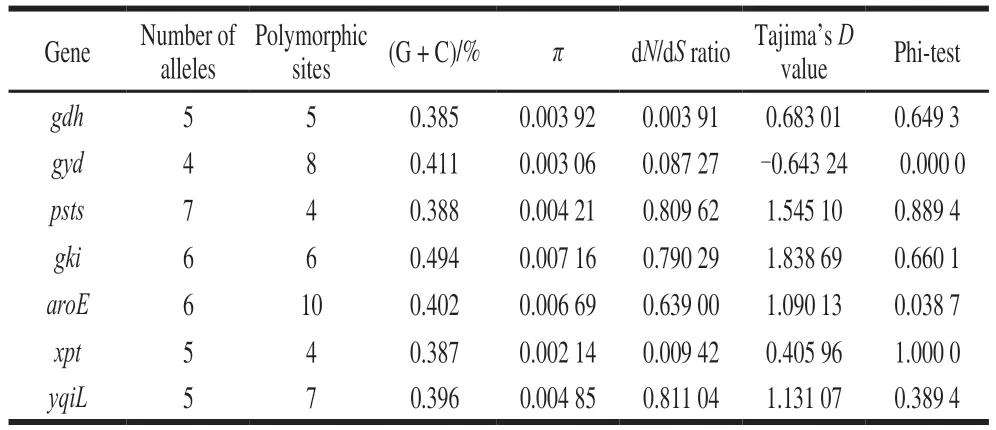
Table 2 Descriptive analysis of the sequence data
TwelveSTswereidentifiedfromthe59E. faecalisisolates(Table3).AmongwhichST-1102,ST-1103,ST-1099,ST-1120andST-1122werenewlyidentifiedinthis study.ST-1103wasasingle-locusvariantofST-21,ST-1120 wasadouble-locusvariantofST-23,ST-1122andST-1099 werebothsingle-locusvariantsofST-71.Themostabundant STwasST-206containing17isolates(28.81%),followedby ST-21containing15isolates(25.42%),ST-466containing7 isolates(11.86%)andST-71containing6isolates(10.17%).TheremainingSTsonlycontained1-4isolates.TheE.faecalisisolatesfromhuman-originwereofSTs1061,466,23,1120,25and206,whileallisolatesfromcoldwaterfish wereofpreviouslyundescribedSTs.TheST-206wasfound inbreastmilk,cheese,camelmilkandmaremilkandST-21 wasfoundincheeseandcoldwaterfish.
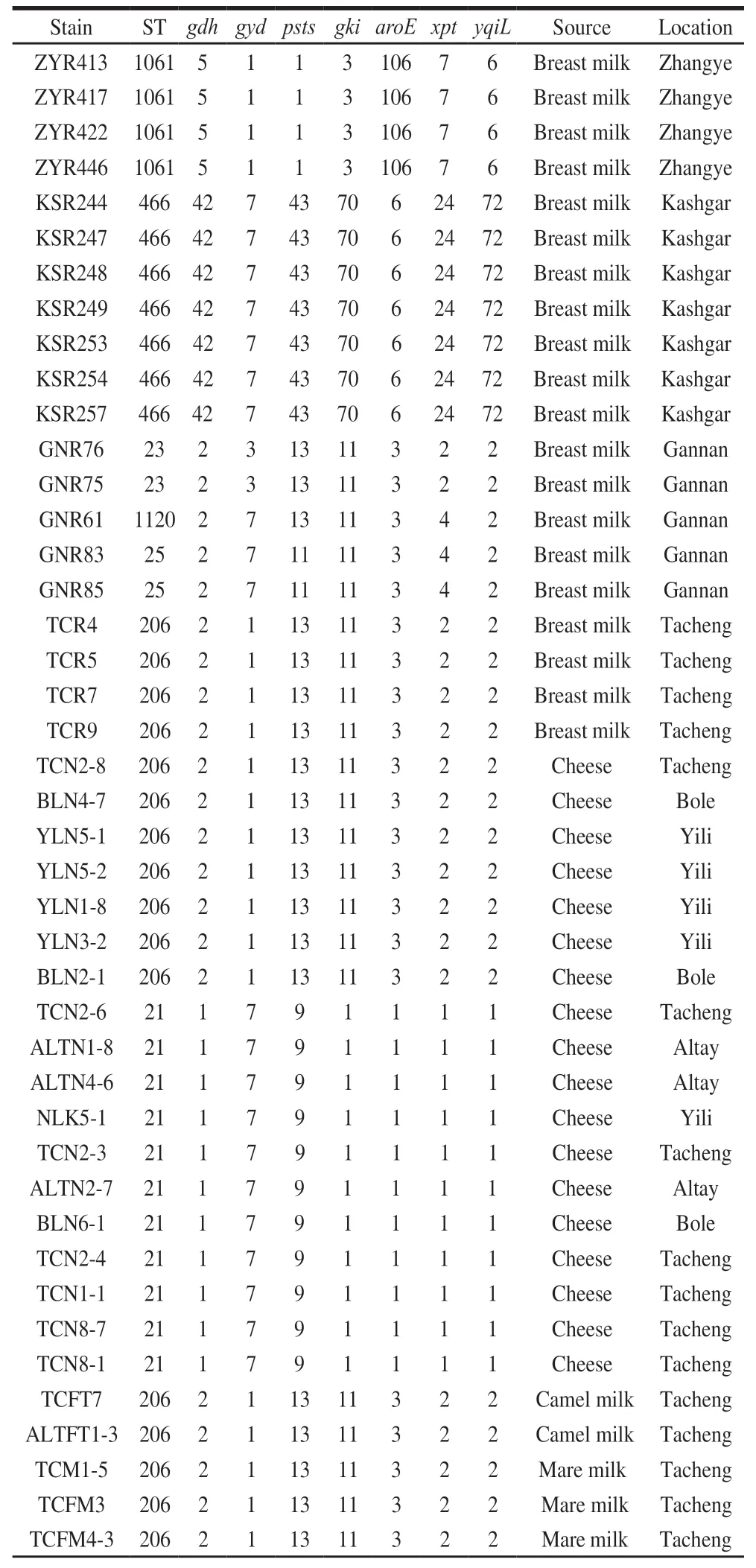
Table 3 STs and allelic profiles of the E. faecalis isolates
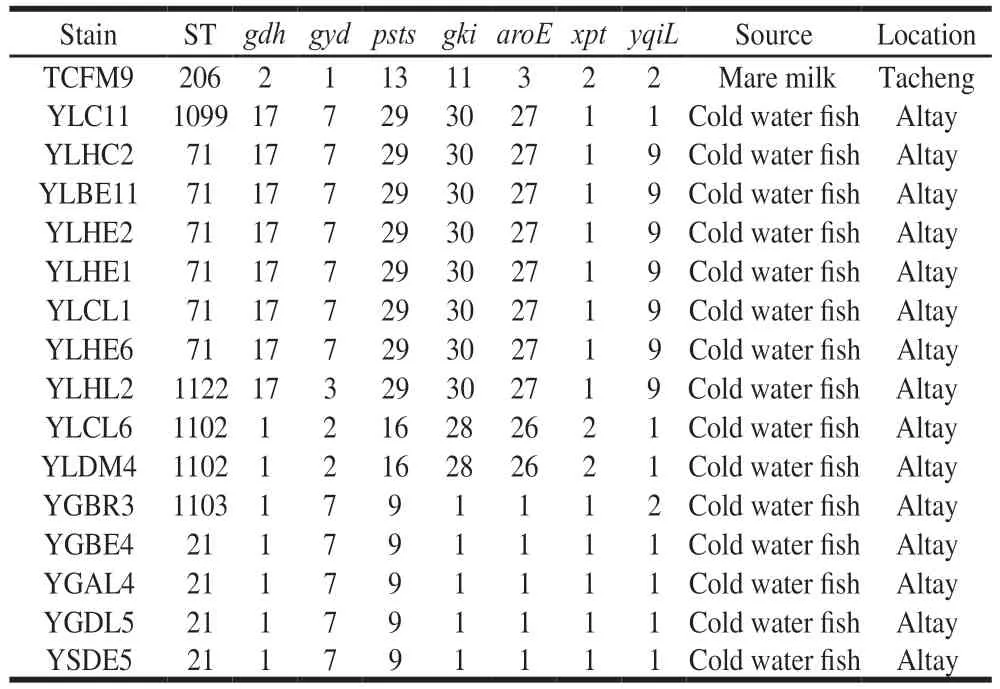
Table 3 (Continued)
2.3 Identification of clonal groups based on STs
WeusedtheeBURSTprogramtoclusterSTsinto groupsbasedonthesimilarityoftheirallelicprofiles.ThisprogramcansubdividelargeMLSTdatasetsinto nonoverlappinggroupsofrelatedSTsorCCsandthen predictthegroupfounder.AswecanseeinFig.2,atotalof 12STsweredividedinto3CCs(CC71,CC25,CC21)and 3singletons.ThethreeCCsincluded44strains,accounting for74.58%ofthetotalstrains.Amongthem,CC25wasthe mostpredominantCCcovered22strainscorrespondingto fourdifferentSTs(ST-23,ST-25,ST-206andST-1120),followedbytheCC71including11strainscorresponding toanotherthreeSTs(ST-71,ST-1099andST-1122).The strainsofCC25werederivedfromcheese,maremilk,camel milkandbreastmilk.Moreover,thestrainsofCC71were allisolatedfromcoldwaterfish.Atthesametime,CC21 containsstrainsnotonlyfromcheesebutalsocold-waterfish.Thesingletonscontained3STtypes,containing13strains,accountingfor22.03%ofthetotal.Ofthese13strains,11 werederivedfrombreastmilkindifferentregions.
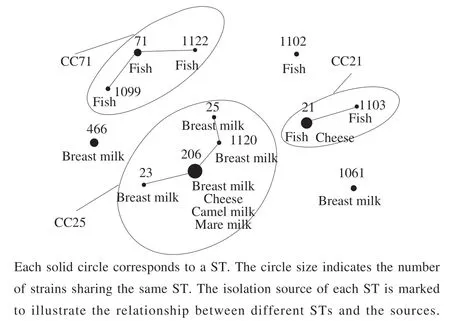
Fig. 2 Population structure of the E. faecalis strains inferred by eBURST analysis
2.4 Comparison analysis of the E. faecalis strains present in different hosts and regions
Geneticrelatednessamongthe59E. faecalisstrains wasachievedwithaphylogenetictreeconstructedfrom concatenatedsequencesusingthemaximum-likelihood method.AsshowninFig.3,clustersii(coldwaterfish),iii(breastmilk1)and v (breastmilk2)hadverylow sequencediversity,whereastheclusters i (cheese)andcluster iv (camelmilk/maremilk/cheese)werehighlyheterogeneous.AllstrainswithinMLSTclusters i (cheese)andmoststrains ofclusters ii(coldwaterfish)incorporatedwithintheCC21 andCC71,respectively.TheMLSTclusters iii(breastmilk1)andcluster iv(camelmilk/maremilk/cheese)onlycontained oneCC(CC21).Moreover,twoSTs(ST-466,ST-1061)were identifiedinclade v (breastmilk2).Althoughconcatenated sequencesclusteringrevealedsomeobviousbreastmilk andcoldwaterfishassociatedlineages,clearlyspecialist subpopulationswerenotfound,asvarioushosttypeswere dispersedacrossdistinctclusters.
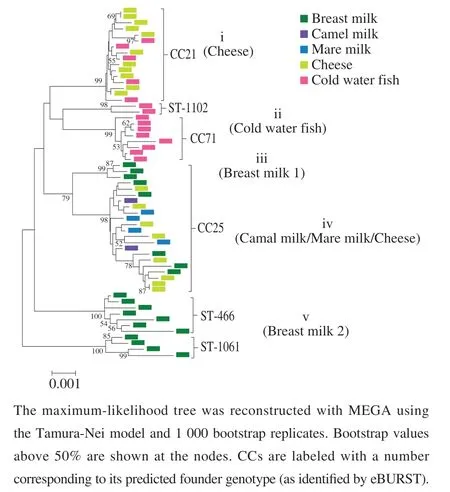
Fig. 3 Phylogenetic analysis of the 59 E. faecalis strains based on the concatenated sequences of seven loci
Aminimum-spanningtreeoftwelveSTswas constructedtoillustratetherelationshipbetweenSTsand differentisolationregions.AsshowninFig.4,the59E. faecalisisolatesweredividedinto2majorbranches.BranchAmainlycontainedisolatesfromAltay(pinkcircles),Kashgar(brickredcircles)andTacheng(orangecircles)inXinjiangUygurAutonomousRegion.BranchBmainly containedisolatesfromGannan(violetcircles)andZhangye(greencircles)inGansuprovince.Nonetheless,someisolates fromGannanwerecloselyrelatedtoisolatesfromthe TachengandYili.Additionally,E. faecalisstrainsfromthe sameregionwerenotallclusteredtogetherwhenviewedas awhole.Forexample,atotalof18strains(ST-21,ST-1099,ST-71,ST-1122,ST-1102,ST-1103)wereretrievedfrom Altayandtheywererelativelydistantfromeachother.The aboveresultsindicatedthatthecompartmentalizationinto subgroupsreflectsadaptationtodifferenthostsratherthan geographicalorigin.
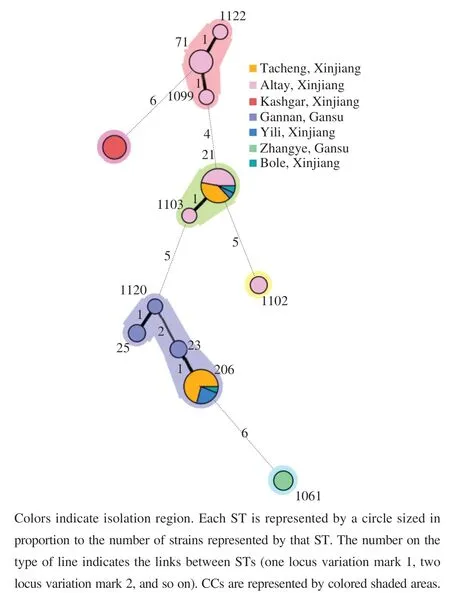
Fig. 4 Minimum-spanning tree analysis of the 59 E. faecalis strains based on MLST data according to geographical origin
2.5 Recombination analysis in E. faecalis
WeusedLIAN-linkagev3.0softwaretoevaluatethe leveloflinkagedisequilibriumbetweenallallelesof59E. faecalisisolates.Thevalues(thestandardizedindex ofassociation)of7genesegmentswas0.6730(P<0.01),whichwassignificantlydifferentfromzero,demonstrating atendencyoflinkagedisequilibriumamongthealleles.In ordertofurtherexploretheeffectofrecombinationonthe populationstructureofE. faecalis,asplitdecomposition analysiswasemployed.Inthisstudy,allgeneshadobvious parallelogramstructures,indicatingthatthesegeneshadbeen affectedbyintergenicrecombinationinevolutionprocess(Fig.5A).Theconcatenatedsequencesofthe7locishowed anapparentnet-likestructure,indicatingthatrecombination eventshadoccurredinthe7locibasedonthephitest(P=0.003468)(Fig.5B).
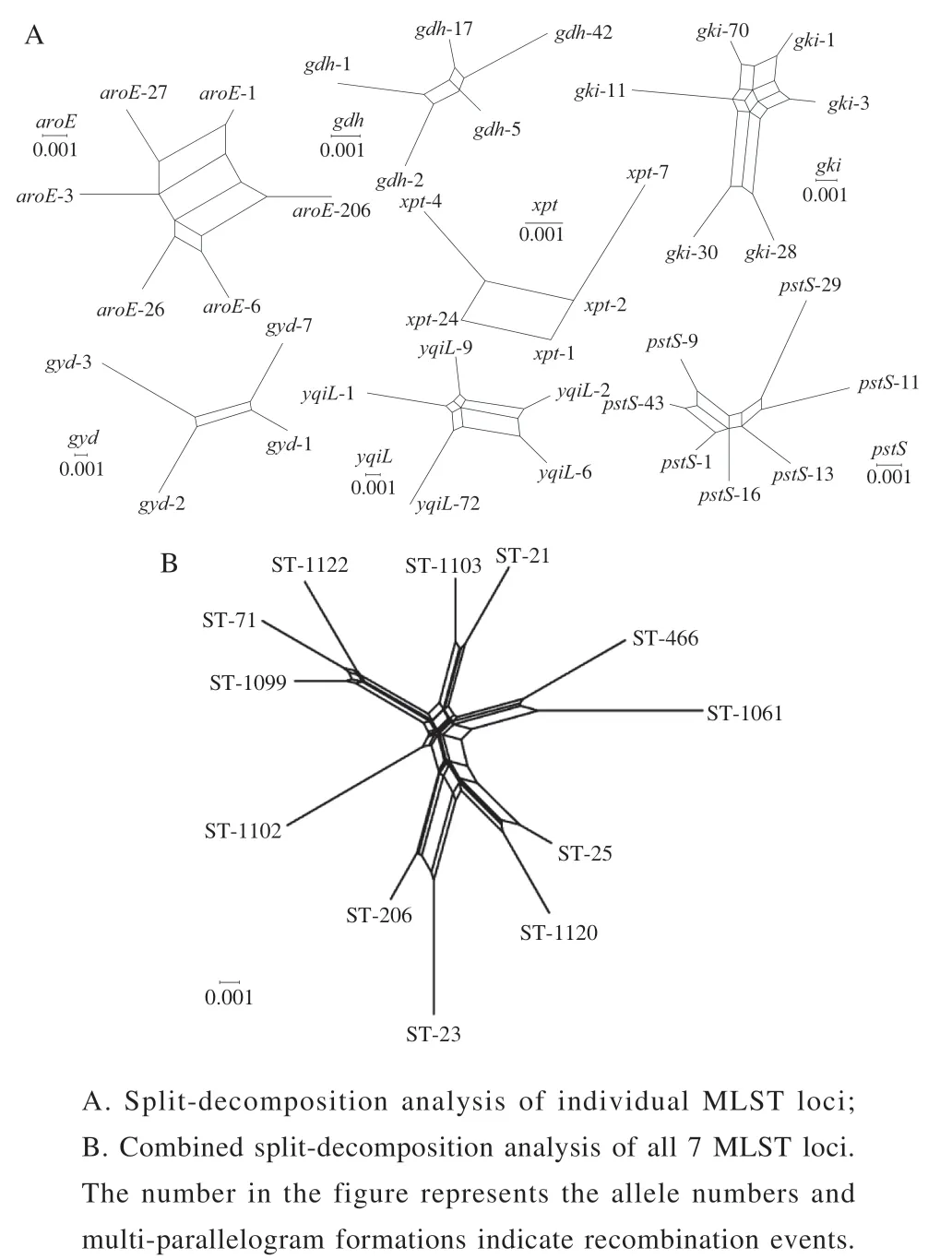
Fig. 5 Split decomposition analysis of the 59 E. faecalis isolates with seven housekeeping gene fragments
3 Discussion
Overthelastthreedecades,E. faecalishasbecomean importantmultidrug-resistantnosocomialpathogen[29].As anecologicalgeneralistmicroorganism,E. faecaliscould spreadfromnosocomialpatientstocommunity-dwelling humans,farmanimalsoranimal-originfood[1,30].Hitherto,a greatdealofresearchhasbeendonetocompareE. faecalisobtainedfromdifferentsources,butmoststudiestypically limitedtonosocomialorigin.Here,wecomparedE. faecalisfromhumanoriginandanimalfoodorigin,andemphasized theroleofhuman-drivendomesticationandinteractionswith livestockprovideopportunitiesforhost-switchingevents betweenhumans,livestockhostsandanimalfoods.
Inourstudy,thepopulationstructureandthe relationshipamongtheisolatesfromdifferentoriginwere analyzedbyMLST.Fifty-ninerepresentativeisolateswere dividedinto12STs,withadiversityof20.34%,which providedalowerdiversitythanisolatesfromothersources.ManyinvestigationsshowedthatE. faecalisexpresshigh geneticdiversity.Inthesestudies,110isolatesofE. faecaliswereassignedto55STswithadiversityofupto50%[28].Subsequently,Leeetal.[31]identified44STsfrom85E. faecalisisolatescollectedfromTraditionalKoreanFermented SoybeanFood,andMohammadietal.[22]identified26STsfrom 90clinicalisolatesinIlam,Iran,withadiversityof51.76%and 28.89%,respectively.Thosedifferencescouldbeduetothe isolatesusedineachstudystemmingfromdifferenthabitats,andvarioushousekeepinggeneswerechosenforanalysis.
EmergingMLSTanalysessuggestedE. faeciumisolatescausingnosocomialinfectionsweregenotypically differentfromcommensalisolatesfromhealthypopulations andisolatesfromdomesticanimals,andtheyclusteredin auniquesubgroup.Thissubgrouphadbeendesignated CC17[13,32-33].TheCC17E. faeciumisolatesexhibitedhighlevelvancomycin,ampicillin,andciprofloxacinresistance andcarriedspecificvirulencegenes[34-35].UnlikeE. faeciumstrains,E. faecalislackadistinctstructureinclades.Some cladesrelatedtooutbreaksinhospitalswerealsofoundin thecommunity.Insomestudies,specificgeneticlineages(CC)ofhospital-adaptedE. faecalisstrainshademerged[28,36].Amongthem,CC2,CC9andCC87werethemostcommon high-riskCCsfoundinhospital-adaptedstrainswhichplayed animportantroleinthespreadofvancomycinresistanceall overtheworld[15,35].Besides,CC16,CC21,CC4andCC40 werethemainsubpopulationsinrecentyears,ofwhichCC16 maybeatetracyclineresistancecluster[19,37-38].Noneofthe STsinourstudybelongedtoCC2,CC9andCC87.However,E. faecalisstrainsisolatedfrombreastmilkbelongingto CC16(ST-1061)werefoundinourstudy,whichwasa concernforhumanhealth.
WefoundnosignificantassociationsbetweenSTs andthesourcesoftheisolates.Theisolatesfromdifferent originsrevealedlowhostspecificityasthesameSTshad beendetectedinvarioushosttypes.ST-206wasshared betweenbreastmilk,cheese,maremilkandcamelmilk.ST-21wassharedbetweencheeseandcoldwaterfish.E. faecalisisconsideredageneralistbacterialspeciesdepicting noprominenthostspecialization[9].TheabilityforE. faecalistojumpintodifferenthosttypesisamajorthreattopublic healthandfoodsecurity.Notably,moresimilarSTswere foundamongcheese,maremilkandcamelmilkandbreast milk.AlmostallSTsofthesethreeoriginswereclusteredin oneCC(CC25).Incontrast,thecold-waterfishstrainswhich wasisolatedfromEerqisiRiverhadacompletelydifferent STsandwasclusteredinCC71(Fig.3).Thoughtheorigin andtransmissionroutesofcloselyrelatedE. faecalisisolates couldnotbeconfirmed,interactionswithhumansordispersal ofanimalsforfoodproductioncannotberuledout[39-41].The domesticationofanimalsandtheintensivelivestockfarming providedmoreopportunitiesforthespreadofE. faecalisstrainsbetweenhumansandanimalfoodasthehuman,animalsandanimalfoodwereinextricablylinked.Human beingsmayrepresentamajorreservoirforthespreadofE. faecalisstrainstootheranimalsandanimalfood.In addition,frequentcommercialtradeinlivinganimalsand animalfoodsmaybeotherreasonsforthespreadofparticular clonestothedifferentregions.
4 Conclusion
Takentogether,all59isolateswereallocatedto12 uniqueSTs,including3CCsand3singletons.Ourfindings suggestthatE. faecalisstrainsshownoprominenthost specializationandvarioushosttypesaredisseminatedacross distinctphylogeneticgroups.Moreover,E. faecalisstrains colonizinghumansandanimalfoodwerecloselyrelated.Theresultsofthestudyalsoemphasizedtheroleofhumandrivendomesticationandinteractionswithlivestockprovide opportunitiesforhost-switchingeventsbetweenhumans,livestockhostsandanimalfoods,complementingprevious reportsmainlyfocusedontheanalysisofclinicalsamples.Therefore,inordertoreducetheriskofzoonoticpotential,ongoingsurveillanceandimprovedinfectioncontrol proceduresatfarmsandherdsmenareindispensable.
