
非变性离子淌度质谱在蛋白质结构及构象疾病研究方面的应用
2022-12-10 06:36:28庄小禹边新宇刘志强宋凤瑞
质谱学报 2022年6期
庄小禹,边新宇,刘 舒,刘志强,宋凤瑞
(1.上海中医药大学科技实验中心,上海 201303;2.中国科学院长春应用化学研究所,长春质谱中心&吉林省中药化学与质谱重点实验室,吉林 长春 130022)
蛋白质为动态实体,要全面了解蛋白质的结构与功能之间的关系,以及蛋白质与配体相互作用对其空间构象的影响,需要能够及时捕捉其构象转变,这对许多“经典”的高分辨结构生物学工具(如X射线晶体学、核磁共振和电子显微镜等)提出挑战。这些方法虽然各具优点,但通常只能提供样品的总体信息,难以对复杂体系中各类蛋白质构象或蛋白质聚集前体进行研究,且无法直接获取关于蛋白质及其复合物相对分子质量及蛋白质复合物结合计量比方面的信息。离子淌度谱(ion mobility spectrometry, IMS)[1]可以分离质量相同而形状不同的离子,与准生理条件下分析和研究生物大分子高级结构、相互作用和动力学的非变性质谱(native mass spectrometry, native MS)联用,不仅可以表征单体蛋白的动态构象,还能够对蛋白复合体功能、结构和组装形式,以及蛋白-配体相互作用进行研究。近年来,由于非变性离子淌度质谱(native ion mobility-mass spectrometry, native IM-MS)可以高灵敏、快速地分析蛋白质混合物中的构象变化,已逐渐成为结构生物学研究的有力工具,并被应用于蛋白质构象、蛋白质-配体相互作用、蛋白质错误折叠、聚集的动力学研究等领域[2-6],为传统的结构生物学研究提供了丰富的互补信息。
1 离子淌度质谱的基本原理及特点
离子淌度谱(IMS)又称离子迁移谱,是根据气相离子在电场和气流形成的漂移区内迁移率不同而分离离子的方法。传统IMS测定的是离子在漂移管中的漂移时间(drift time,tD),不仅取决于离子的质量、带电荷数,还与其大小和形状相关。气相离子进入漂移管后,在电场驱动过程中与惰性漂移气体(通常为氦气、氩气或氮气)不断发生碰撞。通常体积较大的离子比体积较小的离子与漂移管中气体发生碰撞的机会更多,因此迁移得更慢。由于碰撞次数与离子的表面积成正比,IMS测定的漂移时间可用于确定离子旋转时的平均碰撞截面积(collision cross section, CCS),其反映了离子的形状和大小。在蛋白质等生物大分子的结构研究中,离子的碰撞截面积可以直接反映蛋白质的三维形状和复合物堆叠方式。
目前,已有多种离子淌度技术被开发并应用[7-9],较为常见的有漂移时间离子淌度谱(drift-time ion mobility spectrometry, DTIMS)、行波离子淌度谱(travelling-wave ion mobility spectrometry, TWIMS)、捕集离子淌度谱(trapped ion mobility spectrometry, TIMS)、高场非对称离子淌度谱(field-asymmetric ion mobility spectrometry, FAIMS),示于图1。按照电场强度划分,上述前3种可称为低场离子淌度,最后一种为高场离子淌度。使用低场离子淌度的优势是可以确定离子迁移率与CCS之间的关系,因此可以根据实验中测定的漂移时间推导离子的碰撞截面积,这对于研究未知结构的蛋白单体和复合物尤为重要。
除上述几种离子淌度技术,目前正在开发或已投入使用的还有回旋离子淌度、串联离子淌度、超长路径-无损离子淌度。这些新技术都使离子淌度向着高分辨、高灵敏的方向发展。
2 离子淌度质谱可获取的数据及信息形式
2.1 漂移时间
离子的漂移时间是离子淌度谱直接获取的数据形式,反映了离子的碰撞截面积大小。基于离子的漂移时间分离离子,为传统质谱增加了一个分离维度,获得包含离子质荷比 (m/z)、离子丰度和漂移时间的三维信息谱图。因此,IM-MS不仅可以提供结构信息,还可通过将数据分布到第三维来分离重叠峰,从而分析组成高度相似的异质或多分散复合物。
2.2 漂移时间分布
漂移时间分布(arrival time distribution, ATD)峰半高处的漂移时间宽度可以反映蛋白质和复合物的结构异质性,尖而窄的峰表示单一构象,而较宽的峰则表明共存多种状态的构象,溶液中蛋白质组装体的构象多样性。因此,可以通过ATD确定蛋白质相互作用和/或底物结合对所分析蛋白质构象分布的影响。

图1 4种离子淌度技术的基本结构与原理[7]Fig.1 Principle and structure of four main types of ion mobility instruments[7]
2.3 碰撞截面积
通过测定每个电荷状态下的ATD,可以直接或间接计算CCS。CCS也被称为旋转平均碰撞截面积,代表离子与漂移气体碰撞的有效截面积,类似于投影面积。CCS可用于反映离子的结构特征。实验中测得的CCS是分析离子构型的重要评分指标,可以与其他由已知或推测结构计算的CCS进行对比[10]。
2.4 碰撞诱导去折叠
通过对气相离子的碰撞活化作用,可以丰富IM-MS实验的数据量。当需要区分结构上具有细微差异的蛋白质,以及研究蛋白质构象稳定性时,可以通过碰撞诱导去折叠(CIU)实验进行分析。在CIU实验中,逐步升高碰撞能量(collision energy, CE)使蛋白离子活化,并由于库仑斥力的作用诱导蛋白构型发生变化或部分展开,之后经过淌度分离和质谱检测,能够区分同一质荷比下紧凑和伸展的蛋白构象,使用CCS即可判别和量化构象变化[11]。通过CIU指纹图谱可以更清晰直观地观察蛋白活化时的构象变化,不仅可以识别配体或蛋白伴侣键合对蛋白结构的影响,还可以区分源于蛋白多样性(如可变剪接、基因复制、翻译后修饰)的蛋白质异构体。
3 非变性离子淌度质谱在蛋白质结构相关研究领域中的应用
3.1 鉴定单体蛋白折叠结构
蛋白表面的不稳定修饰可以通过动态调控蛋白表面的化学微环境而影响蛋白的局域结构,从而可能介导目标蛋白的特定功能,但一直缺乏合适的结构化学分析手段。Li等[12]利用非变性离子淌度方法,使用非靶向全离子去折叠(AIU)和碎裂(AIF)的方式,高通量无损操控蛋白的气相结构,快速有效剖析糖基化中唾液酸化修饰对转铁蛋白化学、构象、拓扑稳定性的影响。将开发的AIU技术用于对差异唾液酸化修饰的转铁蛋白进行构象稳定性分析,虽然在所有的唾液酸化糖蛋白中都存在多次构象转变,但在相同的活化能范围内,这些转铁蛋白会经历不同的去折叠轨迹。因此,根据不同的3D展开轨迹图可以很好地分辨3种转铁蛋白。
在生物制药领域,离子淌度质谱可在单克隆抗体(mAb)和抗体偶联药物(ADC)等药用蛋白的生产过程中提供完整蛋白的修饰信息和其他特征[13]。如人免疫球蛋白G2(IgG2)具有多种二硫键差异导致的异构体形式,这些异构体具有不同的功能、活性。Bagal 等[14]利用离子淌度质谱解析了这些异构体,并通过分析去糖基化以及二硫键相关位点突变的各种变体,证实了二硫键是引起这些异构体结构差异的关键因素。ADC药物通常存在因小分子药物与抗体偶联位置不同而产生的位置异构体。变性实验条件会破坏ADC子域之间的非共价相互作用,因此半胱氨酰连接的ADC难以通过液相色谱-质谱联用分析抗体比率(DAR)值。Marcoux等[15]将非变性离子淌度谱结合高分辨质谱应用于ADC分析,并给出了指导性建议,包括从去糖基化ADC的非变性离子淌度谱获得药物的平均偶联量和通过计算CCS得到的微小结构变化来区分载药量的差异。对于一些结构差异太细微,不能通过直接比较碰撞截面积(或漂移时间)区分的蛋白质,通过对气相离子的碰撞活化作用,可以丰富离子淌度质谱的实验数据,示于图2。Tian等[16]在对ADC的离子淌度谱分析中引入了CIU方法,成功区分了CCS十分相近的IgGk亚型1~4的mAb。
3.2 应用于蛋白质错误折叠、聚集及与配体相互作用的研究
蛋白质或多肽特定的空间构象对于其发挥正常的生物功能至关重要,结构稳定性的改变会引发蛋白质或多肽的错误折叠及聚集,形成淀粉样原纤维,并最终导致相关疾病的发生[17],如肌萎缩侧索硬化症(ALS)、阿尔茨海默病(AD)、帕金森病(PD)及朊蛋白病等神经退行性疾病和糖尿病(DM)等,这些疾病又称蛋白构象病[18]。而铜锌超氧化物歧化酶(SOD1)、β淀粉样蛋白(Aβ)、突触核蛋白、朊病毒蛋白、人胰淀素(hIAPP)的错误折叠及聚集,分别与ALS、AD、PD、疯牛病及糖尿病等疾病相关。但由于淀粉样原纤维形成过程的中间体具有分散性、多态性和瞬时态的特性,分析和识别这些短暂存在的低聚态中间体一直是一项挑战。
蛋白质通常在原纤维形成之前发生去折叠和错误折叠,因此表征蛋白质的构象特点是研究淀粉样纤维形成的关键问题[19]。IM-MS能够将某一种蛋白单体共存的多种构型(质荷比相同,物理尺寸或形状略有差异)分离开。Zhuang等[20]利用IM-MS及其串联质谱的方法研究了不同溶液体系对SOD1构象的影响,以及SOD1二聚体在气相中的构型变化和分解过程,发现在非变性离子淌度质谱条件下,SOD1二聚体可以保持完整和紧凑构型,在气相中经碰撞活化后构型会逐渐由紧凑变得伸展,同时伴随二聚体解离成单体。该实验共检测到3种构型的二聚体(紧凑、部分伸展、伸展)及2种构型的单体(紧凑和伸展),根据CCS及离子淌度串联质谱实验,确定了SOD1二聚体离子在气相中的3种分解和去折叠途径,示于图3。
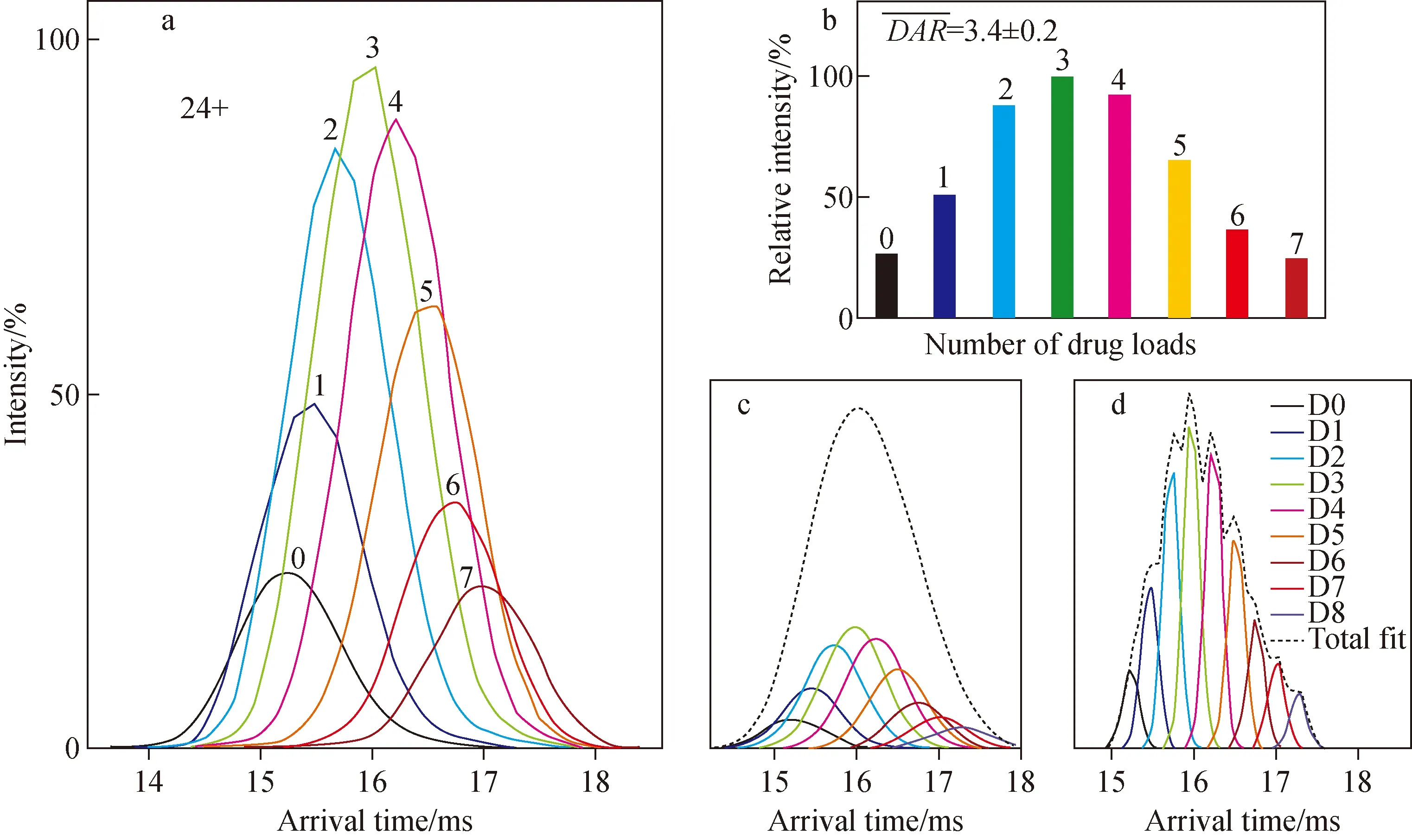
图2 离子淌度质谱法测定去糖基化曲妥珠单抗emtansine的药物-抗体比[15]Fig.2 Determination of the drug-to-antibody ratio (DAR) of deglycosylated trastuzumab emtansined by IM-MS[15]

图3 SOD1二聚体离子[Di]11+(m/z 2 858)的解离及去折叠途径[20]Fig.3 Dissociation and unfolding pathways of SOD1 dimer ion [Di]11+ (m/z 2 858)[20]
此外,由于淀粉样蛋白形成的早期阶段具有高度异质性,多种且快速相互转化的物种共存于溶液中,除研究有聚集倾向的蛋白质单体,识别和表征短暂存在的低聚态中间体是分析淀粉样原纤维形成途径时的难点。非变性离子淌度质谱既可以保留非共价结合的复合物,又可以高灵敏度监测复杂混合样品中低浓度的物种,分离不同的寡聚物而不影响整体平衡,并且能够提供淀粉样蛋白组装方式的信息以及确认有毒低聚体的种类[21]。Illes-Toth等[22]利用IM-MS发现了能够诱导体内聚集的α-突触核蛋白寡聚体的不同构象,虽然低阶低聚物相对非结构化,且主要以线性排列方式结合,但高阶低聚物(如五聚体和六聚体)会形成环状组装体。作者推测这种紧凑的寡聚体可能会促进其他非结构化蛋白质的朊病毒样细胞传播。Leney等[23]比较了β-微球蛋白与它的1个D链单点突变体H51A在体外形成纤维过程的差异,发现H51A会更快地形成高聚体,然而二者所形成低聚体的CCS没有明显差异。近年来,Bowers课题组一直致力于利用离子淌度质谱研究疾病相关的淀粉样蛋白/多肽的聚集过程,如β淀粉样蛋白(Aβ)、突触核蛋白、朊病毒蛋白、人胰淀素样多肽(hIAPP)[24-30]。他们利用IM-MS研究了tau蛋白的PHF核心结构域,证明溶液条件决定了tau蛋白单体和寡聚体的蛋白内和蛋白间相互作用,因此该结构域在tau蛋白的聚集中起决定性作用。通过IM-MS表征tau蛋白的可溶性低聚物,在聚集过程中发现了多达八聚体的可溶性低聚物,这些低聚物会逐渐向不溶性原纤维转移。此外,他们探索了Aβ40和Aβ42的寡聚状态和组装动力学,发现Aβ40主要形成单体和二聚体,同源四聚体丰度较低。因此,推测原纤维是由二聚体形成的四聚体产生的,而缺乏高阶结构限制了Aβ40原纤维的生长。相反,Aβ42形成展开的四聚体,并进一步结合二聚体形成六聚体,2个六聚体相互作用形成有毒的十二聚体[29]。虽然人胰淀素样多肽(hIAPP)与鼠胰淀素样多肽(rIAPP)只有6个氨基酸位点的差异,但rIAPP却不具有聚集倾向。通过离子淌度质谱发现,带有相同电荷数的hIAPP单体具有紧实构象(漂移时间较短)和伸展构象(漂移时间较长),比rIAPP单体多了1个更伸展的构象[30]。分子动力学模拟显示,hIAPP单体的伸展构象由β-发夹结构组成,而rIAPP更紧凑的构象为α-螺旋结构,这与hIAPP纤维形成起始于β-发夹结构中间体的结论接近。
对蛋白质错误折叠疾病的治疗策略研究是在明确蛋白错误折叠、聚集机制的基础之上,通过稳定蛋白质结构,减少、阻止其寡聚体的形成或促进其纤维化,以形成毒性很小或无毒的无定形聚集体等方法来抑制或消除淀粉样蛋白的细胞毒性。Nshanian等[31]利用IM-MS探索了一种镊子分子CLR01抑制tau蛋白聚集的机制,研究显示,tau蛋白与抑制剂结合的化学计量比为1∶1。在CLR01存在的情况下,tau蛋白明显向紧凑构象转变,并且当tau蛋白被磷酸化时效果更加突出,tau蛋白的这种结构重塑抑制了具有聚集能的蛋白质构象异构体的形成。
大量研究发现,各种天然产物可以对淀粉样蛋白的聚集产生显著影响,从而降低聚集诱导的毒性。Zhao等[32]基于离子淌度质谱的碰撞诱导去折叠指纹图谱,证明表没食子IU茶素没食子酸酯(EGCG)能够稳定SOD1二聚体的结构,减缓其去折叠;结合光谱及细胞实验,证明EGCG能够抑制脱金属辅基SOD1(apo-SOD1)的体外聚集,减少聚集过程中疏水区暴露及β层含量的增加,并减少细胞损伤。离子淌度质谱也被用于区分天然小分子配体异构体,以及SOD1与黄酮异构体相互作用的研究[33-34]。Zhuang等[33]利用电喷雾-离子淌度质谱结合光谱方法研究SOD1与黄酮化合物及其异构体之间的非共价相互作用,发现SOD1与黄酮配体结合后其二聚体构型的稳定性得以提升,并根据分子对接方法推测二聚体界面处是黄酮配体主要的结合位点,证实橙皮苷、柚皮苷等黄酮苷类成分具有较好的抑制apo-SOD1体外聚集的能力。此外,该课题组[34]还利用离子淌度质谱研究了白藜芦醇及其衍生物提升SOD1二聚体结构稳定性的作用,示于图4。
SOD1的错误折叠和神经毒性聚集可由突变、金属缺陷和翻译后修饰引起[35]。Bian等[36]
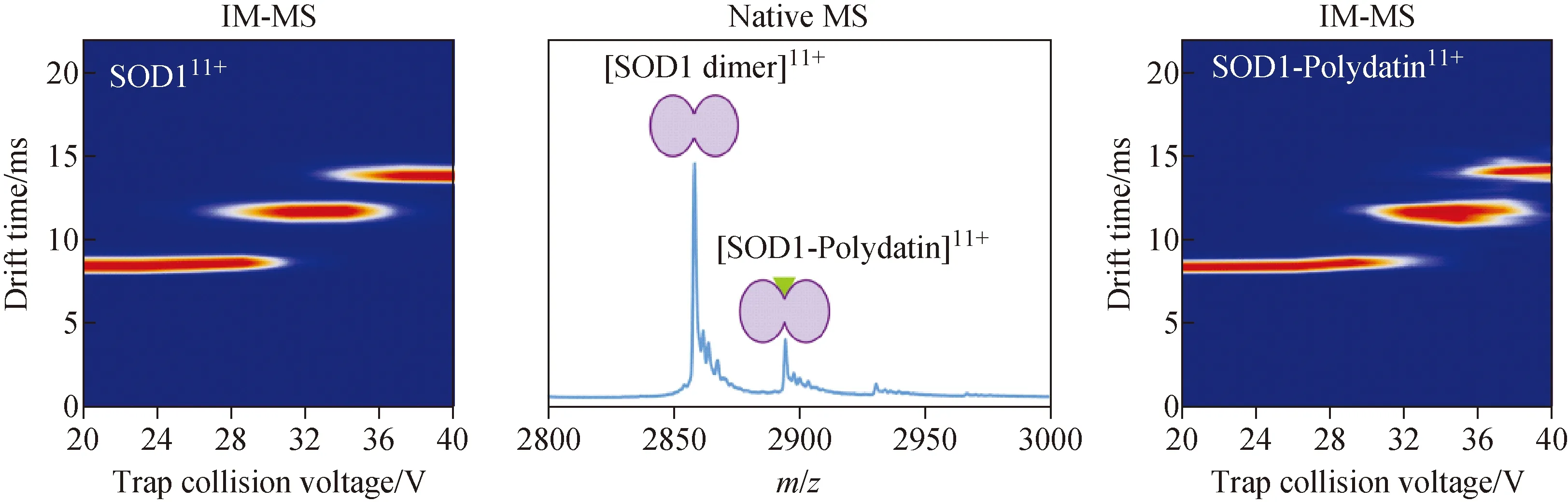
图4 利用native IM-MS研究虎杖苷与SOD1的相互作用[34]Fig.4 Research of interactions between SOD1 and polydatin by native IM-MS[34]
利用非变性离子淌度质谱揭示了锌缺乏SOD1的氧化损伤风险。铜离子在氧化前期被释放,这可能是其结合位点被氧化的结果。另一方面,缺锌SOD1比脱辅基SOD1具有更快的解离倾向。碰撞诱导去折叠表明,缺锌SOD1氧化后更易转变为完全伸展构象。黄豆黄苷能抑制锌缺乏SOD1的氧化,并抑制氧化锌缺乏SOD1构象的转变及聚集,示于图5。
体外研究表明[37-39],EGCG可以改变多种淀粉样蛋白的聚集途径,并将已形成的聚集体解聚,形成无毒性的无定形聚集体。Bleiholder等[39]基于IM-MS方法研究了Aβ淀粉样纤维的形成及其与抑制剂EGCG的相互作用,发现EGCG可有效抑制β构象的寡聚体形成,并将淀粉样纤维转变为紧凑的颗粒状沉淀。Young等[40]证明了IM-MS在筛选和表征Aβ抑制剂方面的能力,确定了小分子与淀粉样蛋白前体(hIAPP)和Aβ-40结合的抑制剂,表征了相互作用的蛋白质种类(单体或寡聚体)并阐明聚集抑制机制,包括抑制剂结合的性质(特异性、非特异性或胶体)、抑制模式-单体结合、单体构象平衡的转变、低聚物的分解等。作者提出IM-MS是一种有前途的高通量筛选工具,并通过IM-MS发现了一种新的hIAPP自组装抑制剂。Pang等[41]利用电喷雾离子淌度质谱结合多种分析方法,分别从hIAPP的构象变化、聚集动力学的影响、聚集中疏水区域的暴露等多方面考察了紫草酸对hIAPP聚集的抑制作用。发现紫草酸与hIAPP结合形成的复合物可以改变hIAPP单体紧实构象和伸展构象的相对丰度,从而维持hIAPP构象的稳定性,并证明紫草酸能够有效抑制hIAPP聚集,减少纤维生成。

图5 利用DTT/H2O2氧化(a,b)和还原(c,d)ApoSOD1(P1)和ApoSOD1(P1)的IM热图,以及P1(e)、P2(f)单体的离子淌度谱图[36]Fig.5 IM heat maps of oxidized (a, b) and reducted (c, d) ApoSOD1 (P1) and ApoSOD1 (P1),IM spectra of +8 charged P1(e) and P2(f) monomer induced by DTT or H2O2[36]
越来越多的研究表明[42-45],某些疾病相关蛋白质聚集过程中形成的可溶性寡聚体具有很强的细胞毒性,可能是致病的重要原因,而蛋白质结构稳定性的降低与寡聚体的形成密切相关。因此,针对疾病相关蛋白的结构稳定性、错误折叠及寡聚体形成与干预措施进行深入研究显得尤为重要。利用灵敏、快捷、特异性强的非变性离子淌度质谱方法,结合多重串联质谱技术(如碰撞诱导解离(CID)以及电子转移解离(ETD)),以及光谱、计算机模拟及细胞生物学实验等技术,针对上述疾病相关蛋白及多肽的结构稳定性、错误折叠及聚集抑制剂的筛选进行深入研究,既有助于阐明蛋白构象病的发病机制,也有利于临床的靶向治疗及相关创新药物的研发。虽然筛选得到的具有稳定蛋白构象、抑制蛋白聚集的天然小分子配体最终应用于临床有待考察,但仍为寻找治疗蛋白质错误折叠疾病的临床药物提供了新途径[46]。
4 结语
非变性离子淌度质谱已逐渐成为一种为传统结构生物学技术提供互补信息的重要研究手段。虽然非变性离子淌度质谱缺少揭示原子水平的分辨率,但分析蛋白质异质复合物及蛋白质-配体相互作用的能力比其他方法更具独特优势。此外,使用离子淌度质谱进行蛋白质结构检测具有快速、灵敏的特点,尤其是近年来将其与native top-down方法进行整合,以及与多种解离方式结合,使非变性离子淌度质谱可以提供更丰富的结构信息。随着高分辨、灵敏便捷的新一代离子淌度质谱技术的不断开发和完善,以及与多样化质谱技术的进一步整合,非变性离子淌度质谱技术在疾病相关的生物大分子结构及相互作用分析方面将拥有更加广阔的应用前景。
猜你喜欢
中国造纸(2022年9期)2022-11-25 02:24:54
环球时报(2019-04-03)2019-04-03 04:15:14
幸福·健康版(2016年10期)2016-11-17 11:21:46
温州大学学报(自然科学版)(2016年1期)2016-10-27 14:57:48
中国卫生标准管理(2015年16期)2016-01-20 09:26:29
西藏科技(2015年10期)2015-09-26 12:10:16
现代检验医学杂志(2015年2期)2015-02-06 02:01:09
现代检验医学杂志(2015年1期)2015-02-06 01:59:20
应用化工(2014年7期)2014-08-09 09:20:23
无机化学学报(2014年5期)2014-02-28 17:31:40
