
伊潘立酮片体外溶出及有区分力溶出曲线的研究
2022-12-09 09:58李大伟刘正平陈倩倩毛楷凡张岱州
食品与药品 2022年6期
李大伟,刘正平,陈倩倩,毛楷凡,张岱州*
(1. 山东省药学科学院 国家药监局药物制剂技术研究与评价重点实验室,山东 济南 250101;2. 山东福瑞达医药集团有限公司 山东省黏膜与皮肤给药技术重点实验室,山东 济南 250101)
伊潘立酮片原研药物由美国Norvatis制药公司研发,Vanda制药公司生产,FDA于2009年5月6日批准上市,商品名为FANAPT[1]。伊潘立酮化学名为4’-[3-[4-(6-氟-1, 2-苯并异噁唑-3-基)哌啶基]丙氧基]-3’-甲氧基苯乙酮[2],分子式为C24H27FN2O4,相对分子质量426.48。伊潘立酮在三氯甲烷或二甲基甲酰胺中易溶,在乙腈中略溶,在甲醇或乙醇中微溶,在水中不溶。
FDA批准上市的FANAPT(Iloperidone Tablets)为口服无包衣片剂,规格分别为1 mg、2 mg、4 mg、6 mg、8 mg、10 mg、12 mg,用于治疗成人精神分裂症的急性发作[3]。根据FDA网站公布的FANAPT申报资料(临床药理学和生物药剂学部分)中披露的信息,伊潘立酮为BCS分类II类,即低溶解性、高渗透性药物。国内目前尚无伊潘立酮片上市。本研究以6 mg片剂为研究对象,研究了原研和自制伊潘立酮片的溶出曲线,并对溶出曲线的区分力进行了研究,以为开展伊潘立酮片的仿制研究提供数据支持。
1 仪器与试药
1.1 仪器
8453型紫外分光光度计(美国Agilent公司);SF-130B型粉碎机(上海天和制药机械有限公司);Retsch PM100型行星高能球磨仪(德国Retsch公司);J-20型气流粉碎机(意大利Tecnologia Meccanica);ZP-21型旋转压片机(上海天和制药机械有限公司);PL203型精密天平,Mettler Toledo AT261型分析天平(梅特勒-托利多公司);Sotax AT 7 smart型自动取样溶出仪(瑞士SOTAX公司);Mastersizer 2000激光粒度仪(英国马尔文仪器有限公司)。
1.2 试药
FANAPT伊潘立酮片(规格:6 mg,批号:V0227A001S4,Vanda Pharmaceuticals Inc.);伊潘立酮对照品(山东省药学科学院,批号:20170915);伊潘立酮原料药(山东省药学科学院,批号:20180305);伊潘立酮片(山东省药学科学院,批号:20180509,20180511,2018051501,2018051502,2018051503,处方编号:1~5);曲拉通,盐酸,结晶乙酸钠,磷酸二氢钾,甲醇(分析纯,国药集团化学试剂有限公司)。
2 方法与结果
2.1 pH值-溶解度曲线测定
按《普通口服固体制剂溶出曲线测定与比较指导原则》[4]中“溶出介质配制方法”配制 pH值分别为 1.0,2.0,3.0,4.0,5.0,6.0,7.0,8.0 的溶出介质。取过量伊潘立酮原料药,气流粉碎,置8支具塞试管中,分别加入各pH值的溶出介质25 ml,置37 ℃水浴振荡过夜,使之形成过饱和溶液,用0.45 μm微孔滤膜过滤,取续滤液,加相应的溶出介质稀释,照紫外-可见分光光度法[5],测定其吸收度,按外标法计算伊潘立酮在不同pH溶液中的溶解度,绘制伊潘立酮在不同pH值条件下的溶解度曲线,结果见图1。

图1 伊潘立酮的pH值-溶解度曲线
2.2 溶出曲线测定
2.2.1 原研片溶出曲线测定 取伊潘立酮原研片6片,按《中国药典》2020年版四部通则中溶出度与释放度测定法第二法(桨法)[5]测定溶出度。经脱气处理的溶出介质的体积为500 ml,转速为50 r/min,水温为37 ℃,在规定的时间点取溶液10 ml(同时补充10 ml 溶出介质),滤过,取续滤液作为供试品溶液。另取伊潘立酮原料药约24 mg,精密称定,置100 ml量瓶中,加无水甲醇约80 ml,超声3 min,加无水甲醇稀释至刻度,摇匀,精密量取5 ml,置100 ml量瓶中,加0.1 mol/L盐酸溶液稀释至刻度,摇匀,作为对照品溶液。照紫外-可见分光光度法[5],以水做空白,在229 nm(溶出介质为0.1 mol/L盐酸溶液、pH 6.8磷酸盐缓冲液和水)或276 nm(溶出介质为 pH 4.5醋酸-醋酸钠缓冲液)处测定对照品溶液和供试品溶液的吸收度,按外标法计算每个时间点每片的溶出度和平均溶出度及相对标准偏差(RSD)。按下式计算溶出度。测定结果见表1。
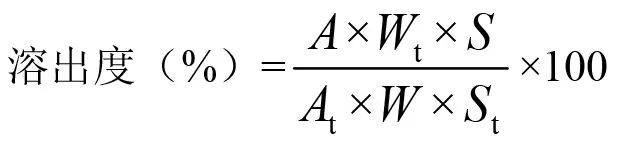
式中,A为供试品溶液的吸光度;Wt为对照品质量;S为供试品溶液的稀释倍数;At为对照品溶液的吸光度;W为供试品标示量;St为对照品溶液的稀释倍数。
由表1可见,原研产品在0.1 mol/L盐酸溶液、pH 4.5醋酸盐缓冲液和pH 6.8磷酸盐缓冲液中,15 min溶出度均达85 %以上,属于快速溶出,其区分力较弱;而在纯化水中,其溶出度在120 min达85 %以上,在180 min达到完全溶出,溶出曲线呈平缓上升趋势,可能具有较好的区分力,需通过试验确认[6-9]。
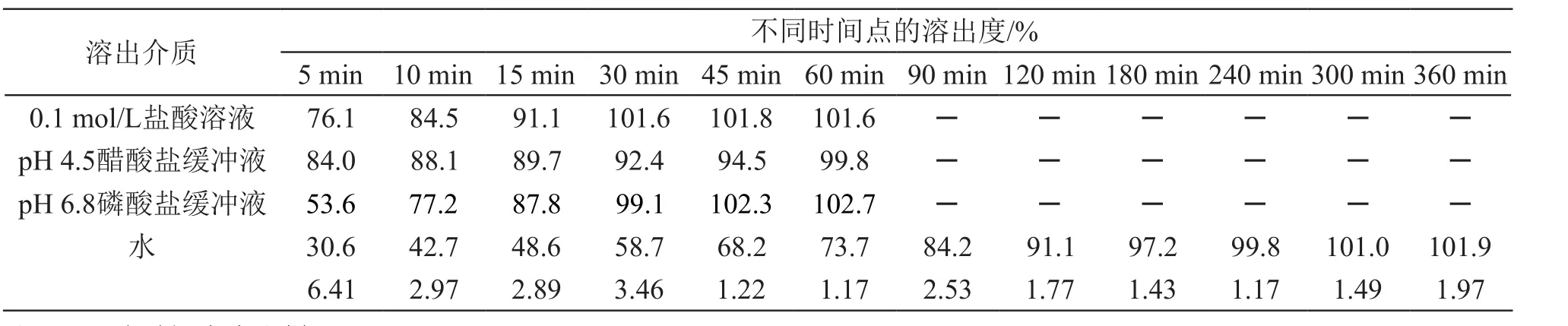
表1 FANAPT(6 mg)的溶出曲线
2.2.2 不同粒度原料药制备片剂溶出曲线的测定 伊潘立酮属BCS分类2类,即低溶解性、高渗透性药物,因此其原料药的粒度为体内吸收的限速步骤。分别采用万能粉碎法、球磨粉碎法、气流粉碎法得到不同粒度的原料药,使用与原研产品相同种类的辅料制备自制片,并测定溶出曲线,以验证各溶出介质的区分力。
2.2.2.1 原料药粒径测定 分散介质:水;分散剂:2 %曲拉通溶液;试验条件:循环速率为220 r/min,超声强度为15 %,超声时间:60 s。
取少量样品,加2 %曲拉通溶液后不断搅拌,待搅拌成混悬液后,当背景正常时,取少量进行测定。遮光度为5 %~20 %,超声60 s后测定。不同粉碎方法处理的原料药粒径测定结果见表2。
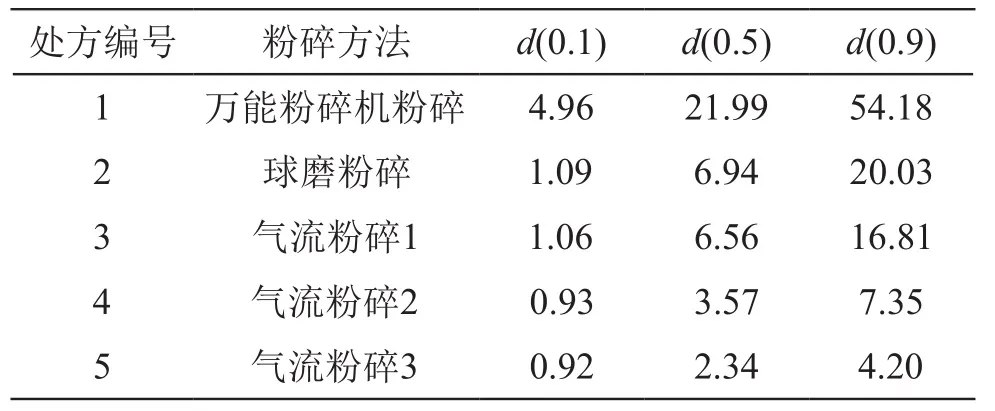
表2 不同方法粉碎后原料药的粒径测定结果/μm
2.2.2.2 自制片溶出曲线测定 按2.2.1项下条件及方法测定不同粒径原料药制备的伊潘立酮自制片溶出曲线,结果见表3。
2.2.2.3 溶出曲线相似性判定[10-11]根据《普通口服固体制剂溶出度实验技术指导原则》,处方1,2,3,4,5及原研产品FANAPT在0.1 mol/L盐酸溶液、pH 4.5醋酸盐缓冲液和pH 6.8磷酸盐缓冲液中15 min时,溶出度均已超过85 %,判定为溶出行为相似。
对各处方在水中的溶出曲线分别与FANAPT进行f2相似因子的计算,当两条溶出曲线f2值不小于50时,可认为溶出曲线相似。
式中,Rt为t时间参比样品平均溶出量;Tt为t时间受试样品平均溶出量;n为取样时间点的个数。
选取时间点为30,60,90,120 min,经计算,各处方的f2值分别为22.6,23.0,40.2,75.9,55.6。由结果可知,各处方在0.1 mol/L盐酸溶液、pH 4.5醋酸盐缓冲液和pH 6.8磷酸盐缓冲液中的溶出曲线相似,无显著差异,而在纯化水中,经f2因子比较,处方1、处方2和处方3均与参比制剂溶出曲线不相似,表明纯化水作为溶出介质可有效区分产品质量,当产品的关键质量属性发生变化时,可有效检出。
3 讨论
仿制药开发以原研产品作为参比制剂,在上市前需进行人体生物等效性试验。由于成本及时间的原因,在开展生物等效性试验之前,一般需要先通过体外评价来确定处方工艺。溶出曲线研究是固体制剂体外等效性评价的重要手段,而溶出曲线的区分力对于保证自研产品与原研产品的质量和疗效一致性尤为关键。根据参考文献[6],具有区分力的溶出曲线应为较平缓上升的曲线,太快和太慢的溶出条件其区分力均不好。根据溶出曲线研究结果,伊潘立酮片在低pH条件下溶出较快,在近中性条件下溶出曲线较为平缓,因此选定纯化水作为溶出介质时,其区分力较好。伊潘立酮为BCS分类2类,即低溶解性、高渗透性,这类药物的粒度对于体内吸收起决定作用。研究中使用不同粒度范围的伊潘立酮制备片剂,其在纯化水介质中的溶出行为存在明显差异。因此,该条件下的体外溶出试验可有效保证产品质量,同时对生物等效性试验具有指导意义。
综上,本研究通过对原研产品进行溶出曲线研究,初步确定了有区分力的溶出条件,并通过制备含不同粒度范围原料药的自制片并测定溶出曲线进行了验证。结果表明,以纯化水为溶出介质,桨法,50 r/min的溶出条件对本品的质量具有良好区分力。
猜你喜欢
药品评价(2020年13期)2020-10-14
工业设计(2020年6期)2020-07-30
中国生殖健康(2019年2期)2019-08-23
中国盐业(2018年20期)2019-01-14
中成药(2018年1期)2018-02-02
中成药(2017年10期)2017-11-16
中成药(2017年10期)2017-11-16
国外医药(抗生素分册)(2016年4期)2016-07-12
特产研究(2016年3期)2016-04-12
中华皮肤科杂志(2014年4期)2014-12-19
